
Male-Biased microRNA Discovery in the Pea Aphid


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA Preparation and Sequencing
2.2. Novel miRNA Identification
2.3. miRNA Distribution Analyses
2.4. miRNA Differential Expression Analyses
2.5. Identification of Potential miRNA Targets
2.6. RNA-Seq and Differential Expression Analyses
3. Results
3.1. RNA Sequencing
3.2. Identification of Novel miRNAs
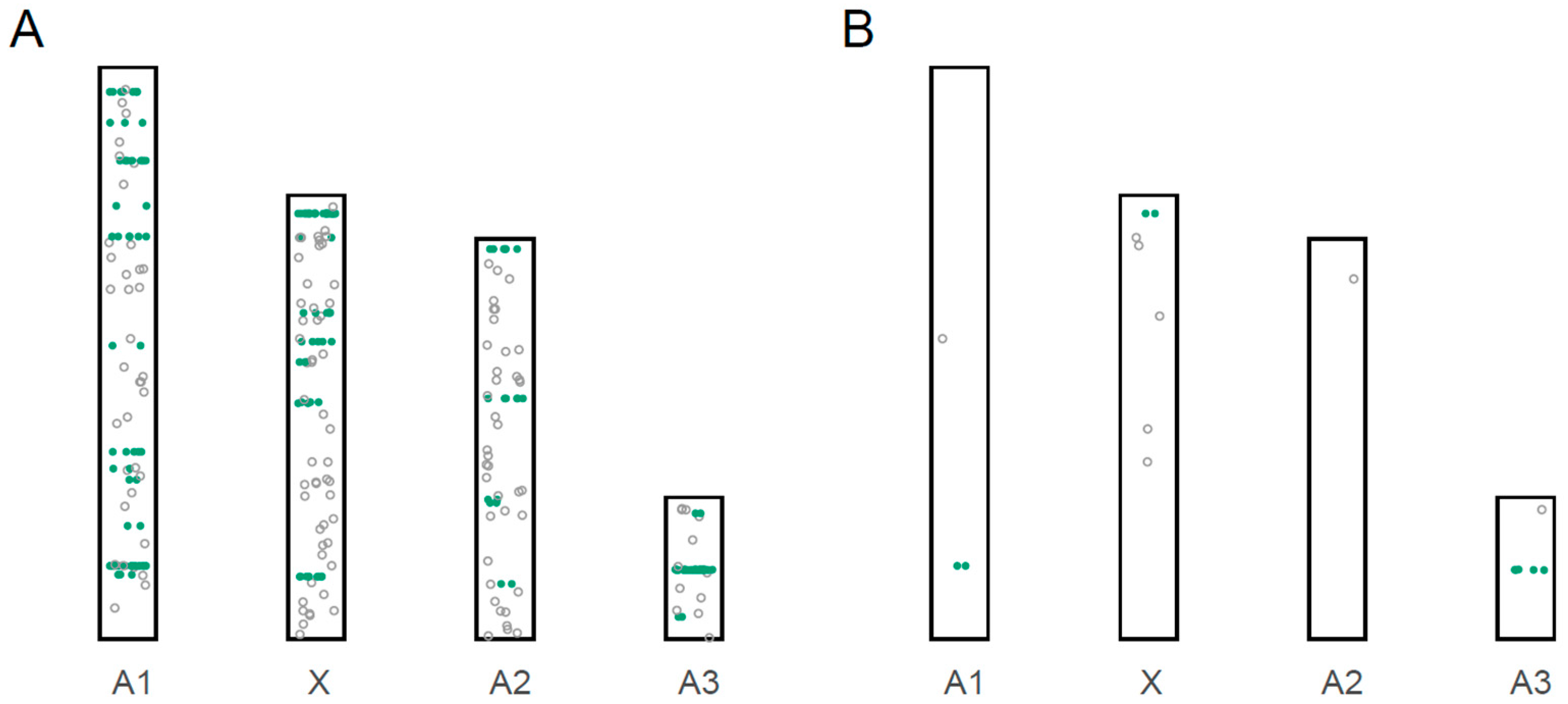
3.3. Localization of miRNA Loci to Chromosomes
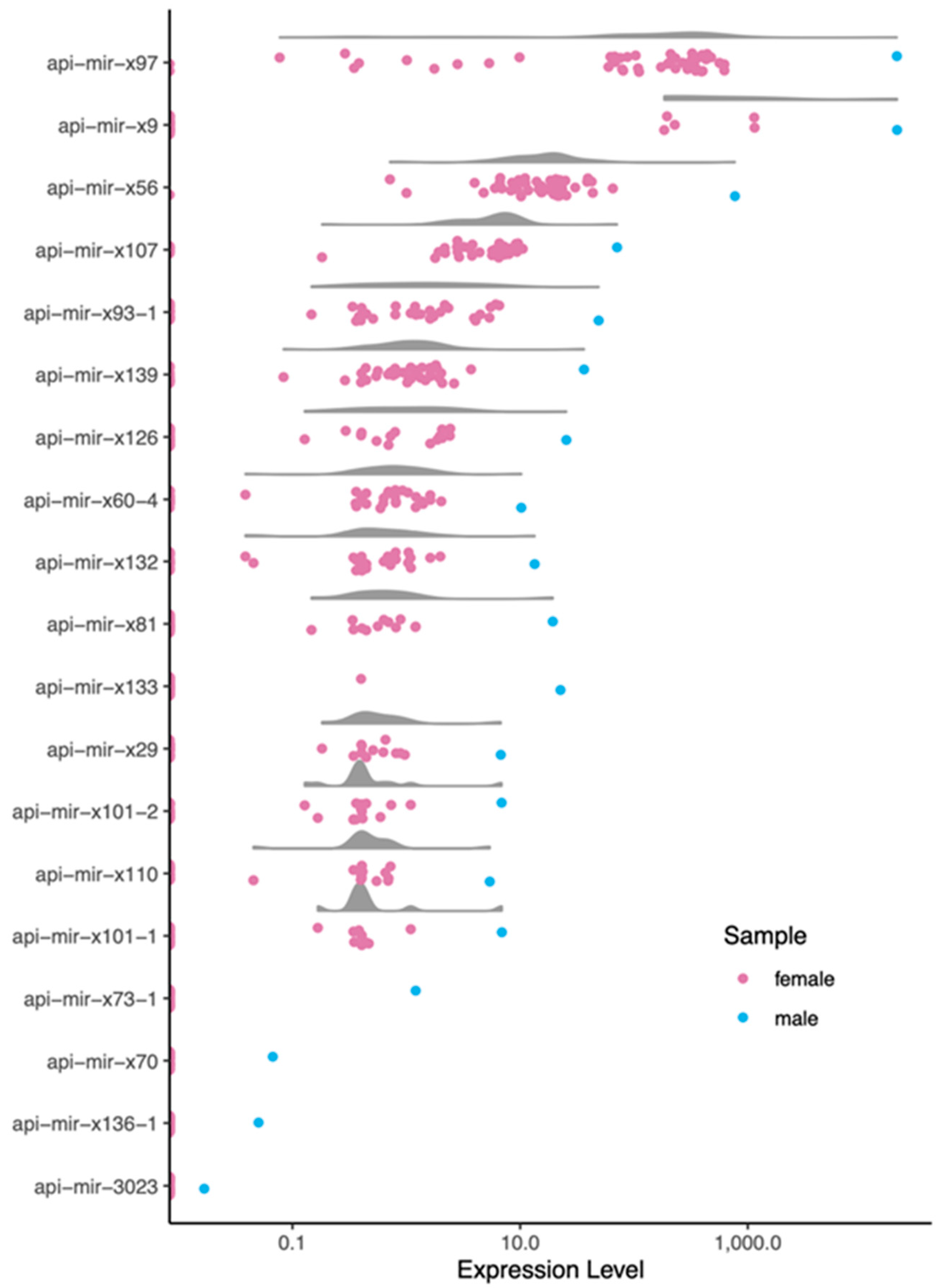
3.4. Male-Biased miRNAs
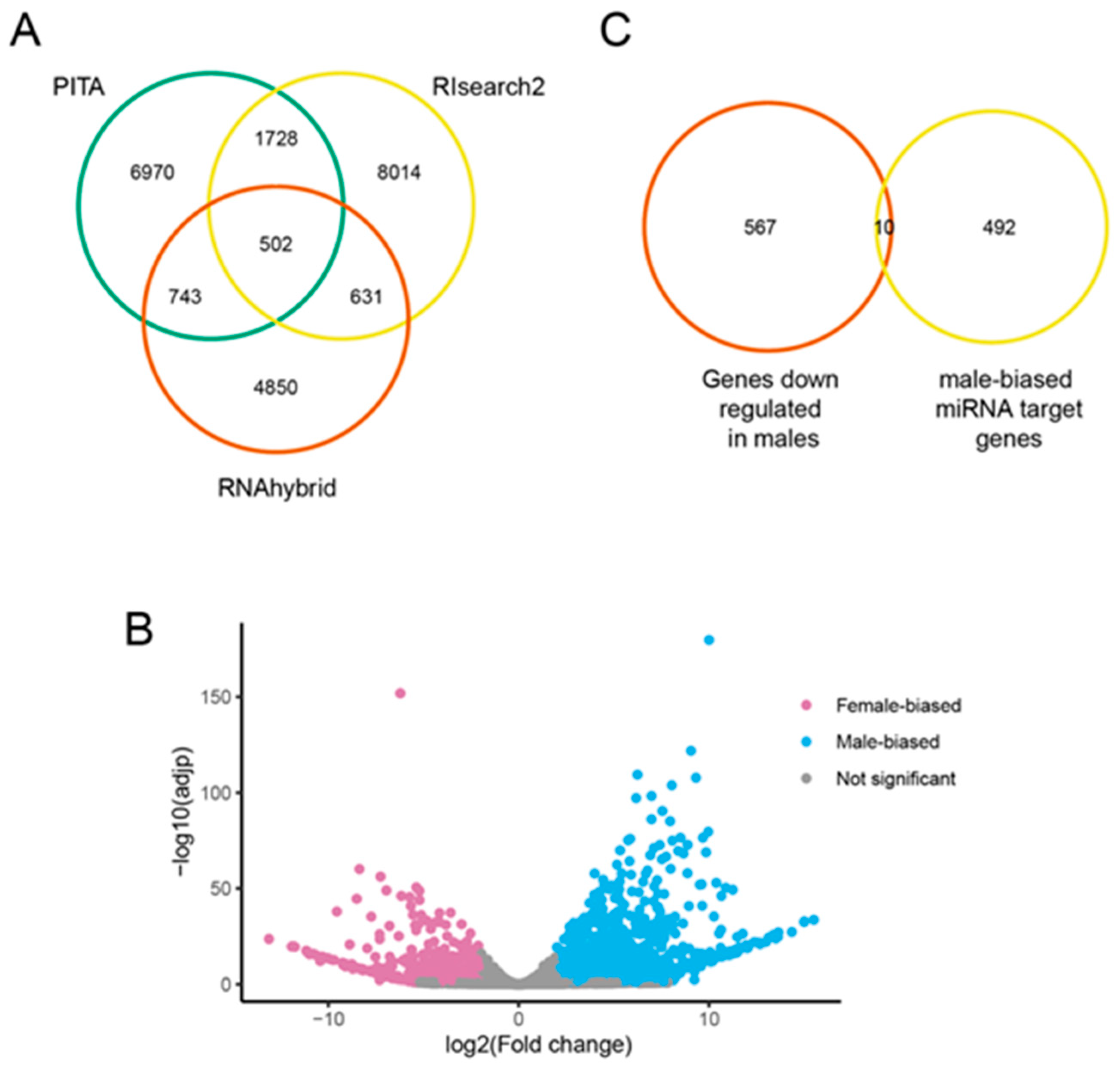
3.5. Predicted Targets of Male-Biased miRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glastad, K.M.; Hunt, B.G.; Goodisman, M.A.D. Epigenetics in Insects: Genome Regulation and the Generation of Phenotypic Diversity. Annu. Rev. Entomol. 2019, 64, 185–203. [Google Scholar] [CrossRef]
- Simpson, S.J.; Sword, G.A.; Lo, N. Polyphenism in Insects. Curr. Biol. 2011, 21, R738–R749. [Google Scholar] [CrossRef]
- Hopkins, B.R.; Kopp, A. Evolution of Sexual Development and Sexual Dimorphism in Insects. Curr. Opin. Genet. Dev. 2021, 69, 129–139. [Google Scholar] [CrossRef]
- Hall, A.B.; Basu, S.; Jiang, X.; Qi, Y.; Timoshevskiy, V.A.; Biedler, J.K.; Sharakhova, M.V.; Elahi, R.; Anderson, M.A.; Chen, X.G.; et al. A male-determining factor in the mosquito Aedes aegypti. Science 2015, 348, 1268–1270. [Google Scholar] [CrossRef]
- Grath, S.; Parsch, J. Sex-Biased Gene Expression. Annu. Rev. Genet. 2016, 50, 29–44. [Google Scholar] [CrossRef]
- Mathers, T.C.; Mugford, S.T.; Percival-Alwyn, L.; Chen, Y.; Kaithakottil, G.; Swarbreck, D.; Hogenhout, S.A.; van Oosterhout, C. Sex-Specific Changes in the Aphid DNA Methylation Landscape. Mol. Ecol. 2019, 28, 4228–4241. [Google Scholar] [CrossRef]
- Bain, S.A.; Marshall, H.; de la Filia, A.G.; Laetsch, D.R.; Husnik, F.; Ross, L. Sex-Specific Expression and DNA Methylation in a Species with Extreme Sexual Dimorphism and Paternal Genome Elimination. Mol. Ecol. 2021. [Google Scholar] [CrossRef]
- Wang, X.; Wheeler, D.; Avery, A.; Rago, A.; Choi, J.-H.; Colbourne, J.K.; Clark, A.G.; Werren, J.H. Function and Evolution of DNA Methylation in Nasonia Vitripennis. PLoS Genet. 2013, 9, e1003872. [Google Scholar] [CrossRef] [PubMed]
- Glastad, K.M.; Gokhale, K.; Liebig, J.; Goodisman, M.A.D. The Caste- and Sex-Specific DNA Methylome of the Termite Zootermopsis Nevadensis. Sci. Rep. 2016, 6, 37110. [Google Scholar] [CrossRef] [PubMed]
- Blackman, R.L. Sex determination in insects. In Insect Reproduction; Leather, S.R., Hardie, J., Eds.; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Richard, G.; Legeai, F.; Prunier-Leterme, N.; Bretaudeau, A.; Tagu, D.; Jaquiéry, J.; Le Trionnaire, G. Dosage Compensation and Sex-Specific Epigenetic Landscape of the X Chromosome in the Pea Aphid. Epigenetics Chromatin 2017, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Gebert, L.F.R.; MacRae, I.J. Regulation of MicroRNA Function in Animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The Impact of MicroRNAs on Protein Output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread Changes in Protein Synthesis Induced by MicroRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Li, X.; Zhang, F.; Coates, B.; Zhang, Y.; Zhou, X.; Cheng, D. Comparative Profiling of MicroRNAs in the Winged and Wingless English Grain Aphid, Sitobion Avenae (F.) (Homoptera: Aphididae). Sci. Rep. 2016, 6, 35668. [Google Scholar] [CrossRef]
- Shang, F.; Niu, J.; Ding, B.-Y.; Zhang, W.; Wei, D.-D.; Wei, D.; Jiang, H.-B.; Wang, J.-J. The MiR-9b MicroRNA Mediates Dimorphism and Development of Wing in Aphids. Proc. Natl. Acad. Sci. USA 2020, 117, 8404–8409. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, L.; Wuchty, S.; Wilson, A.C.C. MicroRNA Regulation in an Ancient Obligate Endosymbiosis. Mol. Ecol. 2018, 27, 1777–1793. [Google Scholar] [CrossRef]
- Sattar, S.; Addo-Quaye, C.; Song, Y.; Anstead, J.A.; Sunkar, R.; Thompson, G.A. Expression of Small RNA in Aphis Gossypii and Its Potential Role in the Resistance Interaction with Melon. PLoS ONE 2012, 7, e48579. [Google Scholar] [CrossRef] [PubMed]
- Legeai, F.; Rizk, G.; Walsh, T.; Edwards, O.; Gordon, K.; Lavenier, D.; Leterme, N.; Mereau, A.; Nicolas, J.; Tagu, D.; et al. Bioinformatic Prediction, Deep Sequencing of MicroRNAs and Expression Analysis during Phenotypic Plasticity in the Pea Aphid, Acyrthosiphon Pisum. BMC Genom. 2010, 11, 281. [Google Scholar] [CrossRef]
- Li, Y.; Park, H.; Smith, T.E.; Moran, N.A. Gene Family Evolution in the Pea Aphid Based on Chromosome-Level Genome Assembly. Mol. Biol. Evol. 2019, 36, 2143–2156. [Google Scholar] [CrossRef] [PubMed]
- Purandare, S.P.; Bickel, R.D.; Jaquiery, J.; Rispe, C.; Brisson, J.A. Accelerated Evolution of Morph-Biased Genes in Pea Aphids. Mol. Biol. Evol. 2014, 31, 2073–2083. [Google Scholar] [CrossRef]
- Jaquiery, J.; Peccoud, J.; Ouisse, T.; Legeai, F.; Prunier-Leterme, N.; Gouin, A.; Nouhaud, P.; Brisson, J.A.; Bickel, R.; Purandare, S.; et al. Disentangling the Causes for Faster-X Evolution in Aphids. Genome Biol. Evol. 2018, 10, 507–520. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, B.; Moran, N.A. The Aphid X Chromosome Is a Dangerous Place for Functionally Important Genes: Diverse Evolution of Hemipteran Genomes Based on Chromosome-Level Assemblies. Mol. Biol. Evol. 2020, 37, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bickel, R.D.; Parker, B.J.; Saleh Ziabari, O.; Liu, F.; Vellichirammal, N.N.; Simon, J.-C.; Stern, D.L.; Brisson, J.A. A Large Genomic Insertion Containing a Duplicated Follistatin Gene Is Linked to the Pea Aphid Male Wing Dimorphism. eLife 2020, 9, e50608. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. MiRDeep2 Accurately Identifies Known and Hundreds of Novel MicroRNA Genes in Seven Animal Clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering MicroRNAs from Deep Sequencing Data Using MiRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The Role of Site Accessibility in MicroRNA Target Recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and Effective Prediction of MicroRNA/Target Duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]
- Alkan, F.; Wenzel, A.; Palasca, O.; Kerpedjiev, P.; Rudebeck, A.F.; Stadler, P.F.; Hofacker, I.L.; Gorodkin, J. RIsearch2: Suffix Array-Based Large-Scale Prediction of RNA–RNA Interactions and SiRNA off-Targets. Nucleic Acids Res. 2017, 45, e60. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expresion Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Park, J.-E.; Heo, I.; Tian, Y.; Simanshu, D.K.; Chang, H.; Jee, D.; Patel, D.J.; Kim, V.N. Dicer Recognizes the 5′ End of RNA for Efficient and Accurate Processing. Nature 2011, 475, 201–205. [Google Scholar] [CrossRef]
- Fagegaltier, D.; König, A.; Gordon, A.; Lai, E.C.; Gingeras, T.R.; Hannon, G.J.; Shcherbata, H.R. A Genome-Wide Survey of Sexually Dimorphic Expression of Drosophila MiRNAs Identifies the Steroid Hormone-Induced MiRNA Let-7 as a Regulator of Sexual Identity. Genetics 2014, 198, 647–668. [Google Scholar] [CrossRef]
- Marco, A.; Kozomara, A.; Hui, J.H.; Emery, A.M.; Rollinson, D.; Griffiths-Jones, S.; Ronshaugen, M. Sex-Biased Expression of MicroRNAs in Schistosoma Mansoni. PLoS Negl. Trop. Dis. 2013, 7, e2402. [Google Scholar] [CrossRef] [PubMed]
- Warnefors, M.; Mössinger, K.; Halbert, J.; Studer, T.; VandeBerg, J.L.; Lindgren, I.; Fallahshahroudi, A.; Jensen, P.; Kaessmann, H. Sex-Biased MicroRNA Expression in Mammals and Birds Reveals Underlying Regulatory Mechanisms and a Role in Dosage Compensation. Genome Res. 2017, 27, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating MicroRNA Annotation and Deep-Sequencing Data. Nucleic Acids Res. 2010, 39, D152–D157. [Google Scholar] [CrossRef]
- Peng, W.; Tariq, K.; Xie, J.; Zhang, H. Identification and Characterization of Sex-Biased MicroRNAs in Bactrocera Dorsalis (Hendel). PLoS ONE 2016, 11, e0159591. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Sex-Chromosome Evolution: Recent Progress and the Influence of Male and Female Heterogamety. Nat. Rev. Genet. 2011, 12, 157–167. [Google Scholar] [CrossRef]
- Jaquiéry, J.; Stoeckel, S.; Rispe, C.; Mieuzet, L.; Legeai, F.; Simon, J.-C. Accelerated Evolution of Sex Chromosomes in Aphids, an X0 System. Mol. Biol. Evol. 2012, 29, 837–847. [Google Scholar] [CrossRef]
- Brisson, J.A.; Nuzhdin, S.V. Rarity of Males in Pea Aphids Results in Mutational Decay. Science 2008, 319, 58. [Google Scholar] [CrossRef][Green Version]
- Despres, L.; David, J.P.; Gallet, C. The Evolutionary Ecology of Insect Resistance to Plant Chemicals. Trends Ecol. Evol. 2007, 22, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Bozzolan, F.; Siaussat, D.; Maria, A.; Durand, N.; Pottier, M.A.; Chertemps, T.; Maïbèche-Coisne, M. Antennal Uridine Diphosphate (UDP)-Glycosyltransferases in a Pest Insect: Diversity and Putative Function in Odorant and Xenobiotics Clearance. Insect Mol. Biol. 2014, 23, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-J.; Marygold, S.J. The UDP-Glycosyltransferase Family in Drosophila Melanogaster: Nomenclature Update, Gene Expression and Phylogenetic Analysis. Front. Physiol. 2021, 12, 648481. [Google Scholar] [CrossRef]
- Fraichard, S.; Legendre, A.; Lucas, P.; Chauvel, I.; Faure, P.; Neiers, F.; Artur, Y.; Briand, L.; Ferveur, J.-F.; Heydel, J.-M. Modulation of Sex Pheromone Discrimination by A UDP-Glycosyltransferase in Drosophila Melanogaster. Genes 2020, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Driscoll, R.M.H.; Grantham, M.E.; Hrcek, J.; Brisson, J.A. Wing Plasticity and Associated Gene Expression Varies across the Pea Aphid Biotype Complex. Evolution 2021, 75, 1143–1149. [Google Scholar] [CrossRef]
- Grantham, M.E.; Shingleton, A.W.; Dudley, E.; Brisson, J.A. Expression Profiling of Winged- and Wingless-Destined Pea Aphid Embryos Implicates Insulin/Insulin Growth Factor Signaling in Morph Differences. Evol. Dev. 2020, 22, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Brisson, J.A. A Laterally Transferred Viral Gene Modifies Aphid Wing Plasticity. Curr. Biol. 2019, 29, 2098–2103.e5. [Google Scholar] [CrossRef]
- Trionnaire, G.; Wucher, V.; Tagu, D. Genome Expression Control during the Photoperiodic Response of Aphids. Physiol. Entomol. 2013, 38, 117–125. [Google Scholar] [CrossRef]
- Richard, G.; Le Trionnaire, G.; Danchin, E.; Sentis, A. Epigenetics and Insect Polyphenism: Mechanisms and Climate Change Impacts. Curr. Opin. Insect Sci. 2019, 35, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, D.G.; Brisson, J.A. Aphids: A Model for Polyphenism and Epigenetics. Genet. Res. Int. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, J.; Zhan, A.; Wang, Y.; Ma, X.; Jie, W.; Cao, Z.; Omar, M.A.A.; He, K.; Li, F. Identification and Analysis of MicroRNAs Associated with Wing Polyphenism in the Brown Planthopper, Nilaparvata Lugens. Int. J. Mol. Sci. 2020, 21, 9754. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome/ Scaffold | Size of Scaffold (bp) | Proportion of Genome | # of Novel miRNAs | # of Previously Described miRNAs | Total # of miRNA | # of Highly Male-Biased miRNAs (5× Higher) |
|---|---|---|---|---|---|---|
| A1 | 170,740,645 | 0.32 | 48 | 51 | 99 | 3 |
| A2 | 119,541,763 | 0.22 | 20 | 37 | 57 | 1 |
| A3 | 42,333,646 | 0.08 | 45 | 9 | 54 | 6 |
| X | 132,544,852 | 0.24 | 72 | 31 | 103 | 7 |
| Unplaced scaffolds | 75,959,697 | 0.14 | 22 | 7 | 29 | 2 |
| Total | 541,120,603 | 1 | 207 | 135 | 342 | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Culbert, E.L.; Brisson, J.A. Male-Biased microRNA Discovery in the Pea Aphid. Insects 2021, 12, 533. https://doi.org/10.3390/insects12060533
Liu X, Culbert EL, Brisson JA. Male-Biased microRNA Discovery in the Pea Aphid. Insects. 2021; 12(6):533. https://doi.org/10.3390/insects12060533
Chicago/Turabian StyleLiu, Xiaomi, Erica L. Culbert, and Jennifer A. Brisson. 2021. "Male-Biased microRNA Discovery in the Pea Aphid" Insects 12, no. 6: 533. https://doi.org/10.3390/insects12060533
APA StyleLiu, X., Culbert, E. L., & Brisson, J. A. (2021). Male-Biased microRNA Discovery in the Pea Aphid. Insects, 12(6), 533. https://doi.org/10.3390/insects12060533