Genome Size Estimation of Callipogon relictus Semenov (Coleoptera: Cerambycidae), an Endangered Species and a Korea Natural Monument

 ,
,
Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Preparation
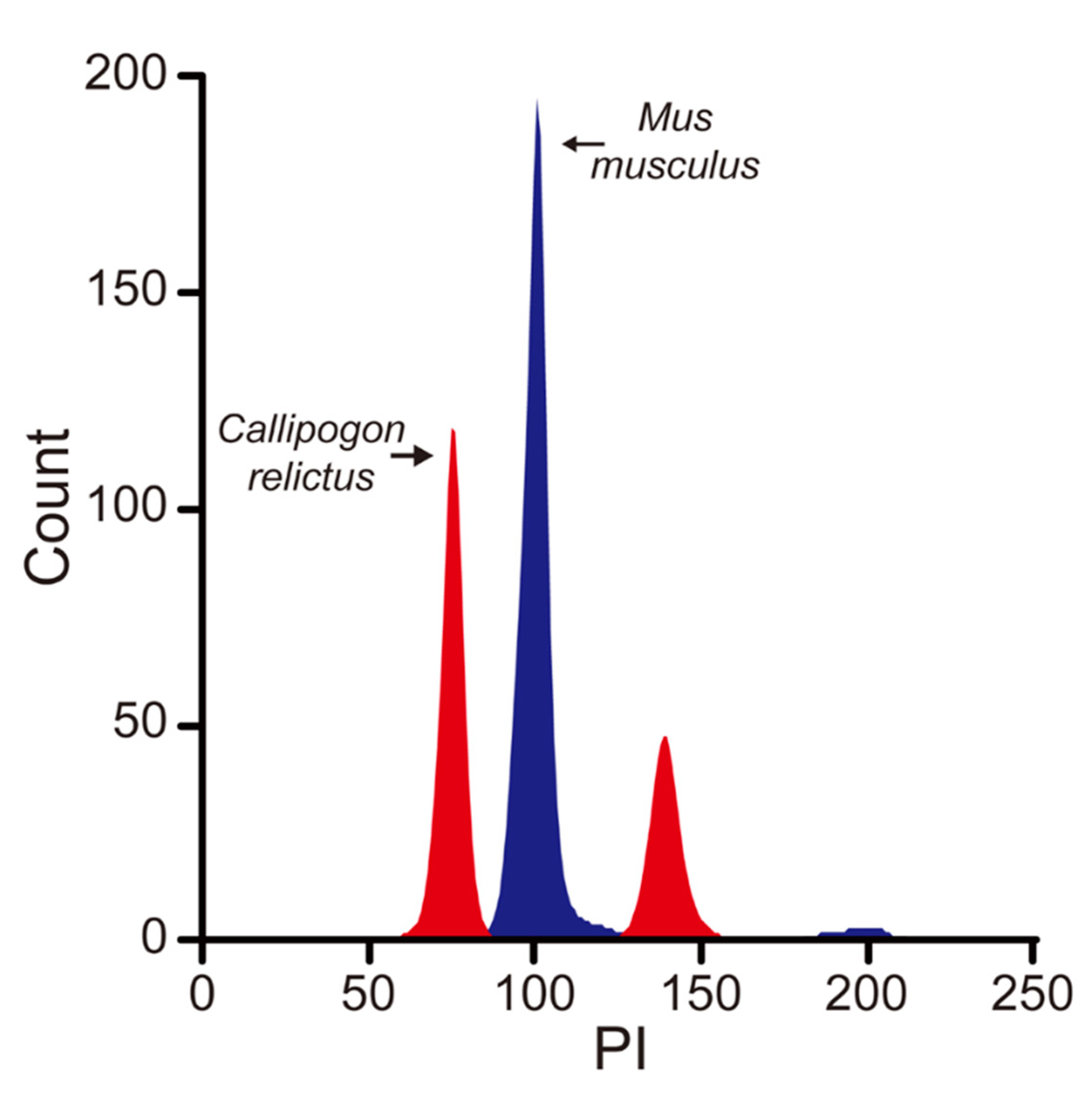
2.2. Genome Size Estimation by Flow Cytometry
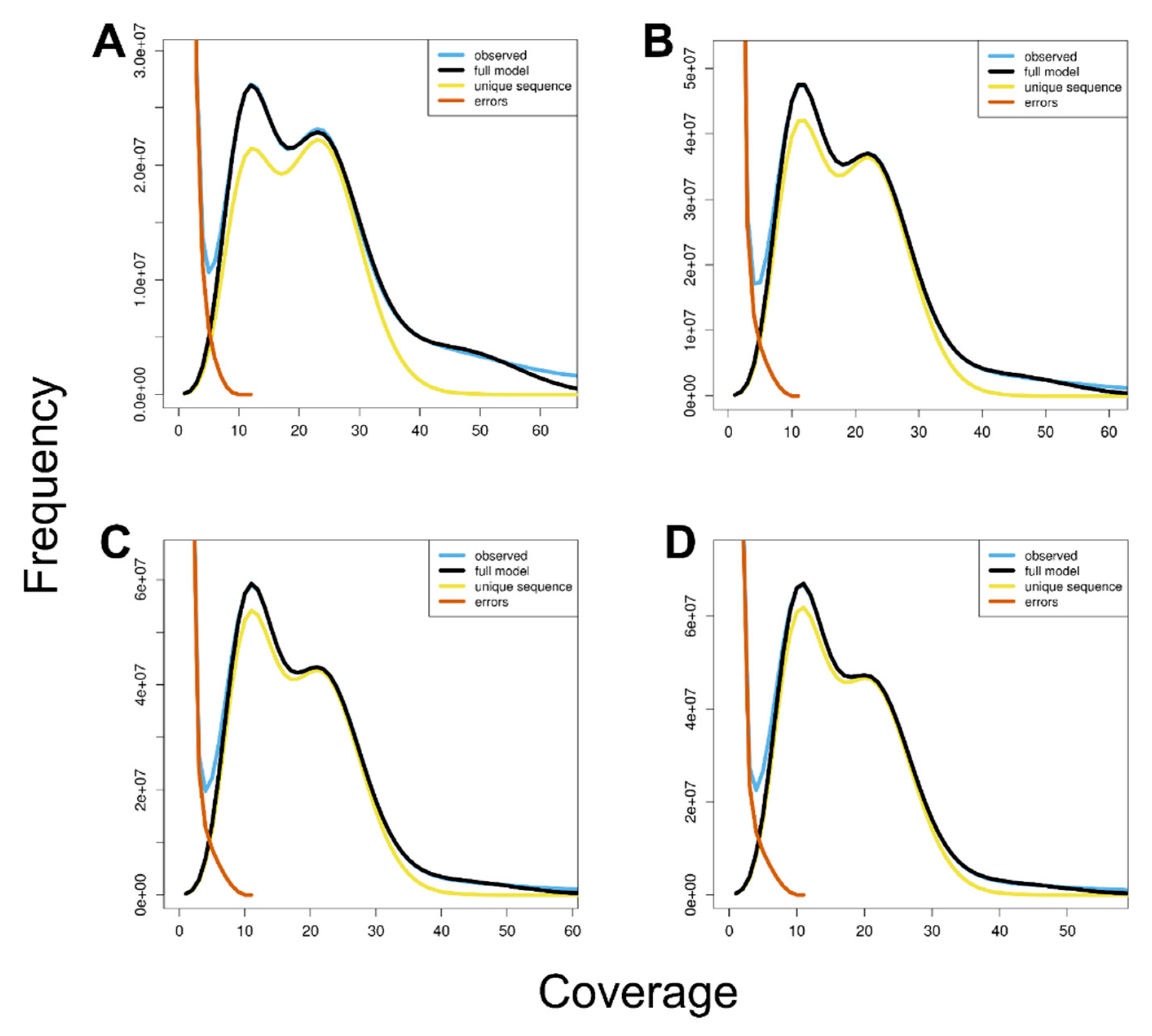
2.3. Genome Size Estimation by k-mer Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Audinet-Serville, J.G. Nouvelle classification de la famille des longieomes. Ann. Soc. Entomol. Fr. 1832, 1, 118–201. [Google Scholar]
- Lameere, A. Révision des Prionides. Neuvième mémoire.–Callipogonines. Ann. Société Entomol. Belg. 1904, 48, 7–78. [Google Scholar]
- Lee, S.-G.; Kim, C.; Kuprin, A.V.; Kang, J.-H.; Lee, B.-W.; Oh, S.H.; Lim, J.J.Z. Survey research on the habitation and biological information of Callipogon relictus Semenov in Gwangneung forest, Korea and Ussurisky nature reserve, Russia (Coleoptera, Cerambycidae, Prioninae). ZooKeys 2018, 792, 45. [Google Scholar] [CrossRef] [PubMed]
- Semenov, A. Callipogon (Eoxenus) relictus sp. no., Vertreter des neotropischen Genus der Cerambyciden in der russischen Fauna. Horae Soc. Entomol. Ross 1898, 32, 562–580. [Google Scholar]
- Kim, S.; de Medeiros, B.A.S.; Byun, B.K.; Lee, S.; Kang, J.H.; Lee, B.; Farrell, B.D. West meets East: How do rainforest beetles become circum-Pacific? Evolutionary origin of Callipogon relictus and allied species (Cerambycidae: Prioninae) in the New and Old Worlds. Mol. Phylogenet Evol. 2018, 125, 163–176. [Google Scholar] [CrossRef]
- Kuprin, A.V.; Bezborodov, V.G. Geographic range of Callipogon relictus Semenov, 1899 (Coleoptera, Cerambycidae) in the Russian Far East. Izv. Akad. Nauk. Ser. Biol. 2012, 459–463. [Google Scholar] [CrossRef]
- Li, J.; Drumont, A.; Xueping, Z.; Meixiang, G.; Wei, Z. The checklist of Northeast Chinas subfamily Prioninae and biological observation of Callipogon (Eoxenus) relictus Semenov-Tian-Shanskij, 1899 (Coleoptera, Cerambycidae, Prioninae). Les Cah. Magellanes 2012, 9, 50–56. [Google Scholar]
- Lee, S.-G.; Kim, C.; Choi, I.-J.; Kuprin, A.V.; Lim, J. A review of host plants of Callipogon (Eoxenus) relictus Semenov (Coleoptera: Cerambycidae: Prioninae), a Korea natural monument, with a new host, Quercus aliena Blume. J. Asia-Pac. Entomol. 2019, 22, 353–358. [Google Scholar] [CrossRef]
- Yi, D.A.; Kuprin, A.V.; Lee, Y.H.; Bae, Y.J. Newly developed fungal diet for artificial rearing of the endangered long-horned beetle Callipogon relictus (Coleoptera: Cerambycidae). Entomol. Res. 2017, 47, 373–379. [Google Scholar] [CrossRef]
- Yin, C.; Shen, G.; Guo, D.; Wang, S.; Ma, X.; Xiao, H.; Liu, J.; Zhang, Z.; Liu, Y.; Zhang, Y.; et al. InsectBase: A resource for insect genomes and transcriptomes. Nucleic Acids Res. 2015, 44, D801–D807. [Google Scholar] [CrossRef]
- Li, F.; Zhao, X.; Li, M.; He, K.; Huang, C.; Zhou, Y.; Li, Z.; Walters, J.R. Insect genomes: Progress and challenges. Insect Mol. Biol. 2019, 28, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Doležel, J.; Greilhuber, J.; Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2007, 2, 2233. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.C.; Dong, Z.W.; He, J.W.; Zhao, R.P.; Wang, W.; Li, X.Y. Genome size of 14 species of fireflies (Insecta, Coleoptera, Lampyridae). Zool. Res. 2017, 38, 449–458. [Google Scholar] [CrossRef][Green Version]
- Wu, C.; Twort, V.G.; Crowhurst, R.N.; Newcomb, R.D.; Buckley, T.R. Assembling large genomes: Analysis of the stick insect (Clitarchus hookeri) genome reveals a high repeat content and sex-biased genes associated with reproduction. BMC Genom. 2017, 18, 884. [Google Scholar] [CrossRef] [PubMed]
- Hartke, J.; Schell, T.; Jongepier, E.; Schmidt, H.; Sprenger, P.P.; Paule, J.; Bornberg-Bauer, E.; Schmitt, T.; Menzel, F.; Pfenninger, M.; et al. Hybrid Genome Assembly of a Neotropical Mutualistic Ant. Genome Biol. Evol. 2019, 11, 2306–2311. [Google Scholar] [CrossRef]
- Zhang, S.; Gu, S.; Ni, X.; Li, X. Genome Size Reversely Correlates With Host Plant Range in Helicoverpa Species. Front. Physiol. 2019, 10, 29. [Google Scholar] [CrossRef]
- Hanrahan, S.J.; Johnston, J.S. New genome size estimates of 134 species of arthropods. Chromosome Res. 2011, 19, 809–823. [Google Scholar] [CrossRef]
- Hare, E.E.; Johnston, J.S. Genome size determination using flow cytometry of propidium iodide-stained nuclei. Methods Mol. Biol. 2011, 772, 3–12. [Google Scholar] [CrossRef]
- Manekar, S.C.; Sathe, S.R. Estimating the k-mer Coverage Frequencies in Genomic Datasets: A Comparative Assessment of the State-of-the-art. Curr. Genom. 2019, 20, 2–15. [Google Scholar] [CrossRef]
- Simpson, J.T. Exploring genome characteristics and sequence quality without a reference. Bioinformatics 2014, 30, 1228–1235. [Google Scholar] [CrossRef]
- Austin, C.M.; Tan, M.H.; Harrisson, K.A.; Lee, Y.P.; Croft, L.J.; Sunnucks, P.; Pavlova, A.; Gan, H.M. De novo genome assembly and annotation of Australia’s largest freshwater fish, the Murray cod (Maccullochella peelii), from Illumina and Nanopore sequencing read. Gigascience 2017, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Heckenhauer, J.; Frandsen, P.B.; Gupta, D.K.; Paule, J.; Prost, S.; Schell, T.; Schneider, J.V.; Stewart, R.J.; Pauls, S.U. Annotated Draft Genomes of Two Caddisfly Species Plectrocnemia conspersa CURTIS and Hydropsyche tenuis NAVAS (Insecta: Trichoptera). Genome Biol. Evol. 2019, 11, 3445–3451. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Herrero, J.F.; Frias-Lopez, C.; Escuer, P.; Hinojosa-Alvarez, S.; Arnedo, M.A.; Sanchez-Gracia, A.; Rozas, J. The draft genome sequence of the spider Dysdera silvatica (Araneae, Dysderidae): A valuable resource for functional and evolutionary genomic studies in chelicerates. Gigascience 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Pflug, J.M.; Holmes, V.R.; Burrus, C.; Johnston, J.S.; Maddison, D.R. Measuring genome sizes using read-depth, k-mers, and flow cytometry: Methodological comparisons in beetles (Coleoptera). Genes Genomes Genet. 2020, 10. [Google Scholar] [CrossRef]
- Schoville, S.D.; Chen, Y.H.; Andersson, M.N.; Benoit, J.B.; Bhandari, A.; Bowsher, J.H.; Brevik, K.; Cappelle, K.; Chen, M.M.; Childers, A.K.; et al. A model species for agricultural pest genomics: The genome of the Colorado potato beetle, Leptinotarsa decemlineata (Coleoptera: Chrysomelidae). Sci. Rep. 2018, 8, 1931. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Guo, L.T.; Wang, S.L.; Wu, Q.J.; Zhou, X.G.; Xie, W.; Zhang, Y.J. Flow cytometry and K-mer analysis estimates of the genome sizes of Bemisia tabaci B and Q (Hemiptera: Aleyrodidae). Front. Physiol. 2015, 6, 144. [Google Scholar] [CrossRef]
- He, K.; Lin, K.; Wang, G.; Li, F. Genome Sizes of Nine Insect Species Determined by Flow Cytometry and k-mer Analysis. Front. Physiol. 2016, 7, 569. [Google Scholar] [CrossRef]
- Gregory, T.R. Animal Genome Size Database. Available online: http://www.genomesize.com (accessed on 26 August 2020).
- Lim, J.; Kim, M.; Kim, I.-K.; Jung, S.; Lim, J.-S.; Park, S.-Y.; Kim, K.-M.; Kim, C.; Byun, B.-K.; Lee, B.-W. Molecular identification and larval description of Callipogon relictus Semenov (Coleoptera: Cerambycidae), a natural monument of South Korea. J. Asia-Pac. Entomol. 2013, 16, 223–227. [Google Scholar] [CrossRef]
- Lim, J.; Yi, D.-K.; Kim, Y.H.; Lee, W.; Kim, S.; Kang, J.-H.; Kim, I.-K. Complete mitochondrial genome of Callipogon relictus Semenov (Coleoptera: Cerambycidae): A natural monument and endangered species in Korea. Mitochondrial DNA Part B 2017, 2, 629–631. [Google Scholar] [CrossRef]
- Steiner, C.C.; Putnam, A.S.; Hoeck, P.E.; Ryder, O.A. Conservation genomics of threatened animal species. Annu. Rev. Anim. Biosci. 2013, 1, 261–281. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insert size (bp) | 550 |
| Total raw reads | 596,389,120 |
| Total raw sequences (bp) | 60,235,301,120 |
| Average length of raw reads (bp) | 101 |
| Total trimmed reads | 561,403,836 |
| Total trimmed sequences (bp) | 55,811,494,971 |
| Average length of trimmed reads (bp) | 99.4 |
| Reads filtered out (%) | 5.87 |
| Sequences filtered out (%) | 7.34 |
| Contents | 17-mer | 19-mer | 21-mer | 23-mer |
|---|---|---|---|---|
| Estimated genome size (bp) * | 1,750,660,507 | 1,786,300,918 | 1,831,421,762 | 1,882,948,731 |
| Estimated genome size ** | ||||
| Heterozygosity (%) | 1.71 | 1.81 | 1.77 | 1.70 |
| Genome haploid length (bp) | 1,517,383,829 | 1,562,628,029 | 1,589,446,896 | 1,611,360,578 |
| Genome repeat length (bp) | 1,096,296,131 | 883,450,205 | 796,477,101 | 757,352,211 |
| Genome unique length (bp) | 421,087,698 | 679,177,824 | 792,969,795 | 854,008,367 |
| Model fit (%) | 98.46 | 99.46 | 99.47 | 99.39 |
| Read Error Rate (%) | 0.15 | 0.23 | 0.25 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.-S.; Jin, S.; Cho, N.; Lim, J.; Kim, C.-H.; Lee, S.-G.; Kim, S.; Park, J.-S.; Kim, K.; Park, C.; et al. Genome Size Estimation of Callipogon relictus Semenov (Coleoptera: Cerambycidae), an Endangered Species and a Korea Natural Monument. Insects 2021, 12, 111. https://doi.org/10.3390/insects12020111
Yu Y-S, Jin S, Cho N, Lim J, Kim C-H, Lee S-G, Kim S, Park J-S, Kim K, Park C, et al. Genome Size Estimation of Callipogon relictus Semenov (Coleoptera: Cerambycidae), an Endangered Species and a Korea Natural Monument. Insects. 2021; 12(2):111. https://doi.org/10.3390/insects12020111
Chicago/Turabian StyleYu, Yun-Sang, Soyeong Jin, Namjoon Cho, Jongok Lim, Cheol-Hak Kim, Seung-Gyu Lee, Sangil Kim, Jong-Seok Park, Keekwang Kim, Chungoo Park, and et al. 2021. "Genome Size Estimation of Callipogon relictus Semenov (Coleoptera: Cerambycidae), an Endangered Species and a Korea Natural Monument" Insects 12, no. 2: 111. https://doi.org/10.3390/insects12020111
APA StyleYu, Y.-S., Jin, S., Cho, N., Lim, J., Kim, C.-H., Lee, S.-G., Kim, S., Park, J.-S., Kim, K., Park, C., & Cho, S.-J. (2021). Genome Size Estimation of Callipogon relictus Semenov (Coleoptera: Cerambycidae), an Endangered Species and a Korea Natural Monument. Insects, 12(2), 111. https://doi.org/10.3390/insects12020111