
Assay Optimization Can Equalize the Sensitivity of Real-Time PCR with ddPCR for Detection of Helicoverpa armigera (Lepidoptera: Noctuidae) in Bulk Samples

 ,
,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
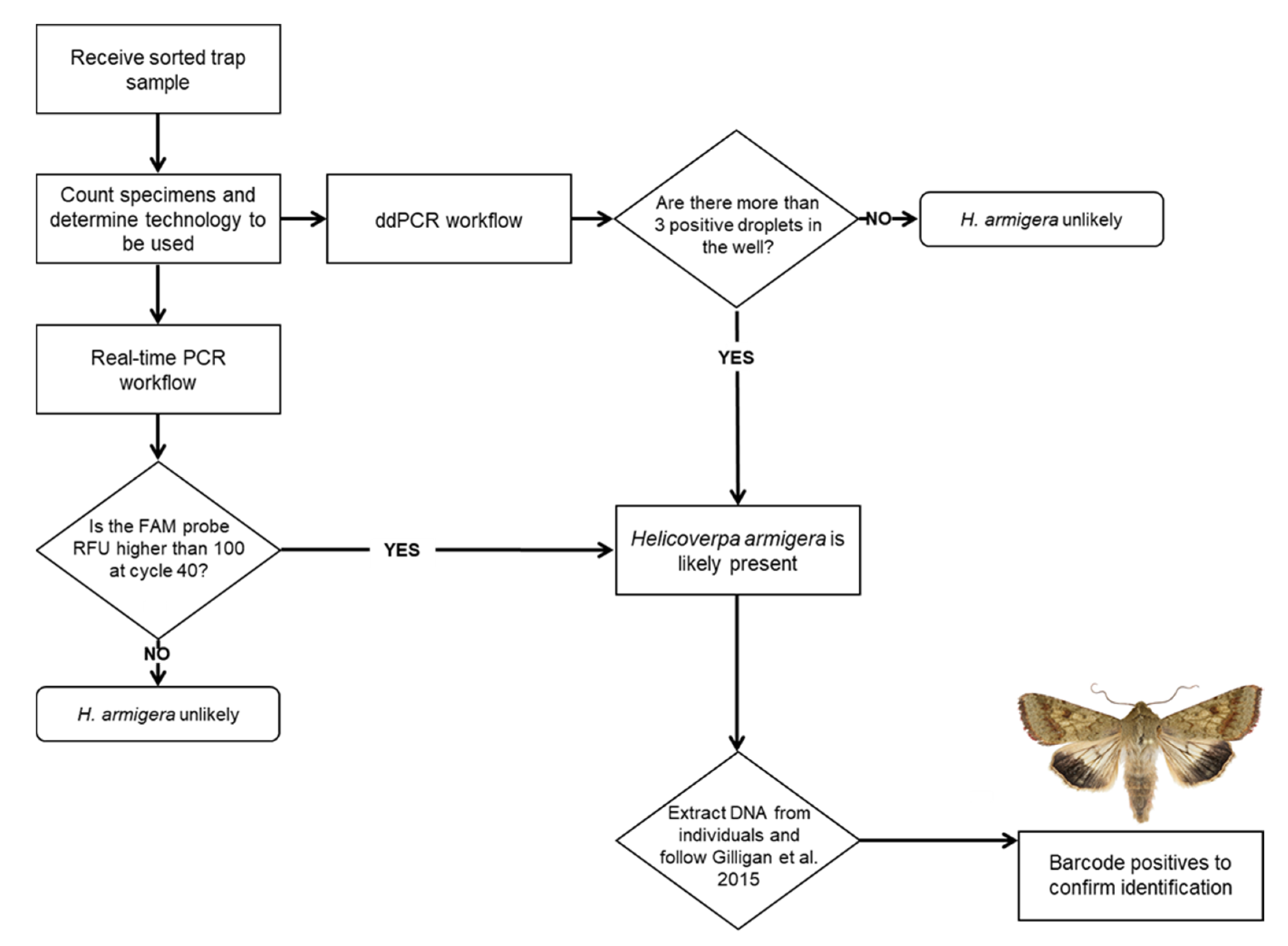
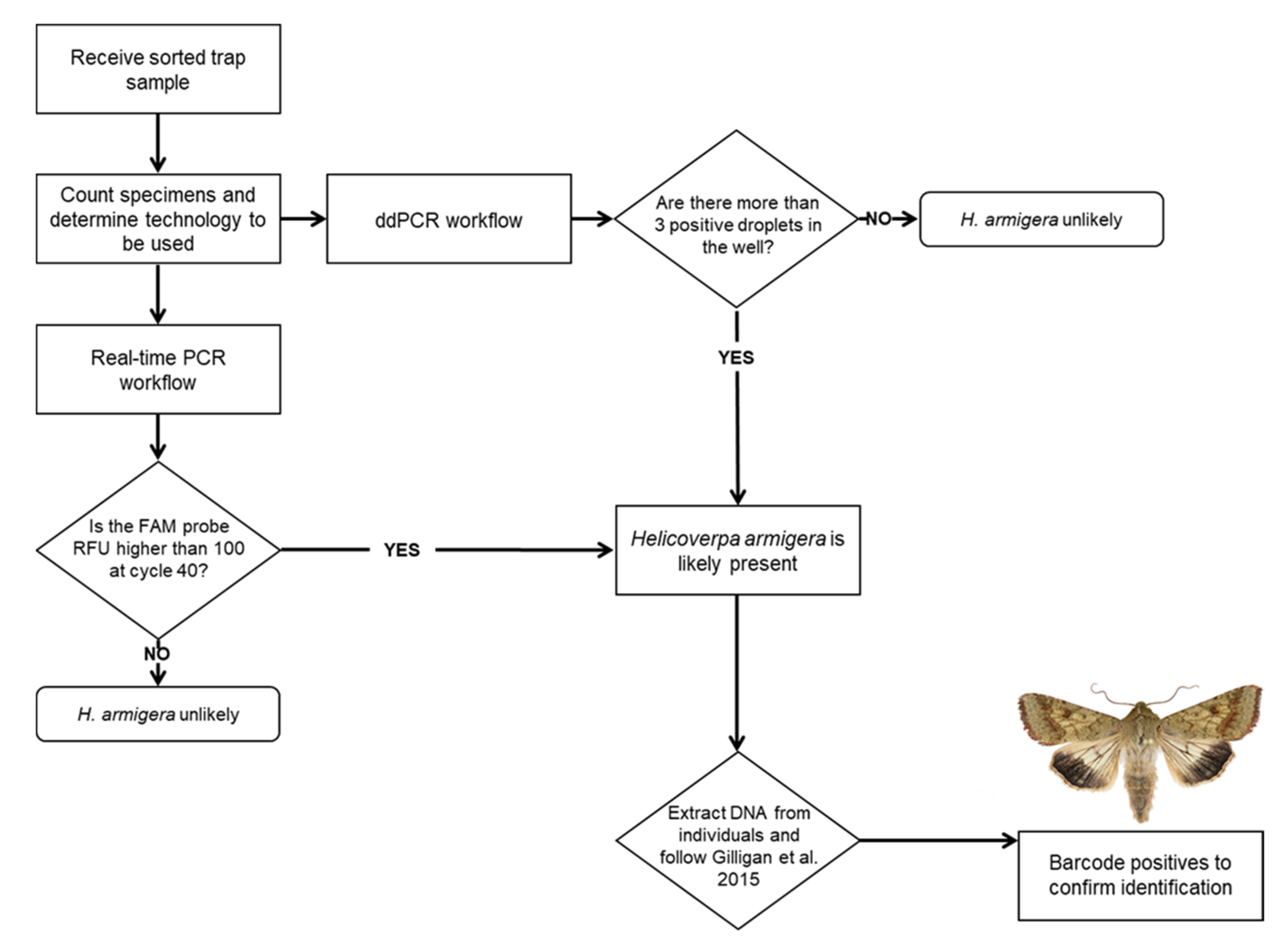
2. Materials and Methods
2.1. Origin and Type of Insect Material
2.2. DNA Extraction from Individual Specimens
2.3. Bulk DNA Extraction
2.4. Primer and Probe Design
2.5. Real-Time PCR
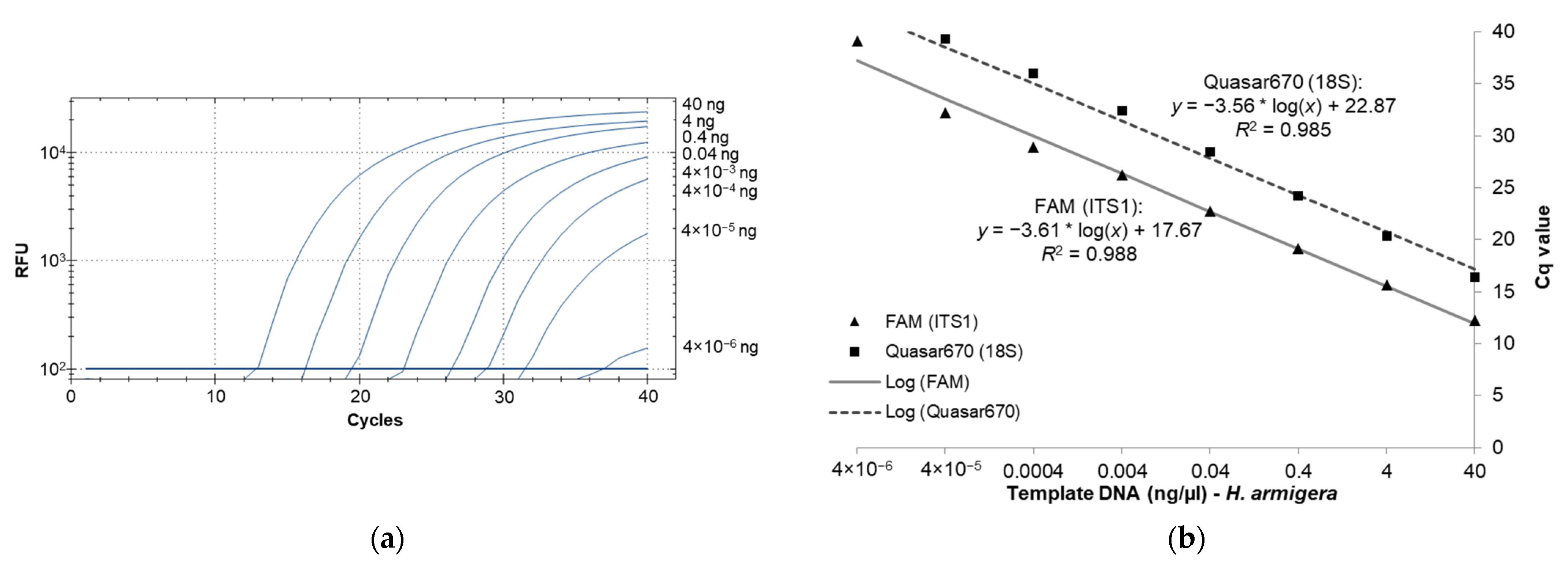
2.6. Real-Time PCR Sensitivity Analyses
- yi = Cq observed value referring to the i-th dilution;
- β0 = intercept;
- β1 = slope;
- Xi = the i-th dilution associated to the observed value yi;
- ei = residual associated to the yi observation.
2.7. Real-Time PCR Comparative Analyses
- yijk = RFU observed value referring to the k-th bulk sample of combination of the i-th level of use of 18S control factor with the j-th level of use of bead purification factor;
- µ = intercept;
- wk = effect of k-th bulk samples in the observed value yijk;
- αi = effect of i-th level of the use of control factor in the observed value yijk;
- βj = effect of j-th level of the use of bead purification factor in the observed value yijk;
- αβij = effect of the interaction of the i-th level of the use of control factor as the j-th level of the use of bead purification factor;
- eijk = residual associated with the yijk observation;
2.8. ddPCR
3. Results
3.1. Real-Time PCR Assay Optimization
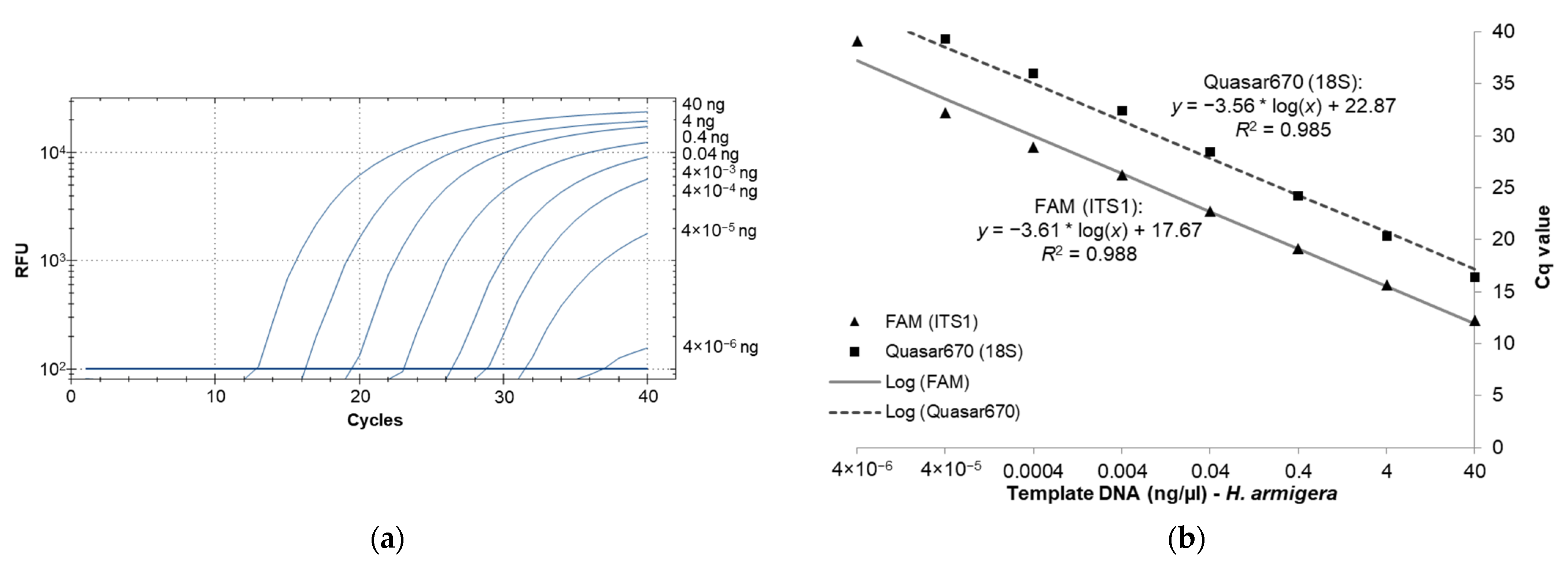
3.2. Real-Time PCR Sensitivity as Determined with High-Quality Template DNA from H. armigera
3.3. Increased Salt Improves Squish Buffer Bulk Extractions
3.4. Real-Time PCR Bulk Sample Results
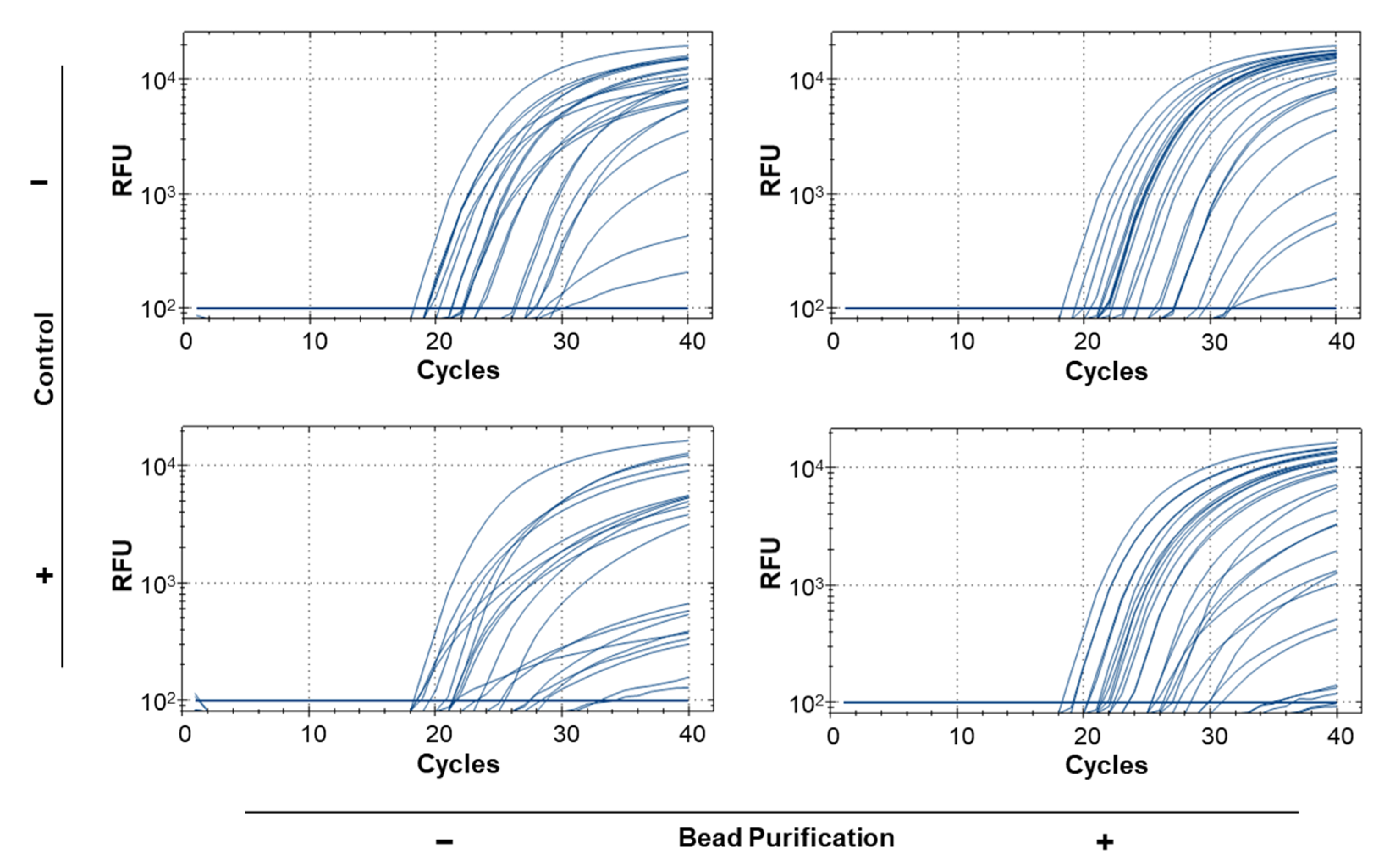
3.5. Bead Purification and Simplex Reactions Improve Sensitivity for Real-Time PCR Using Bulk Samples
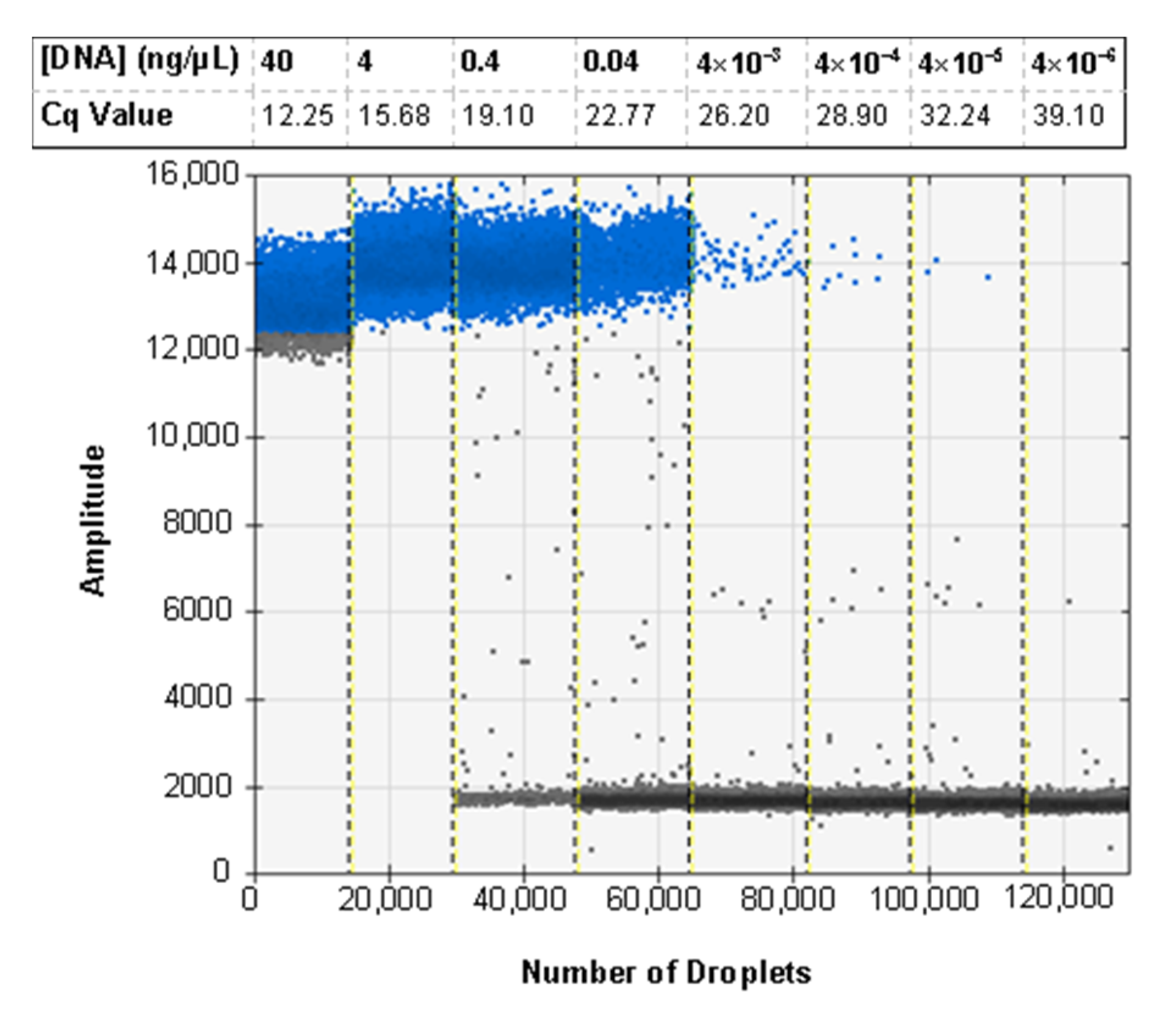
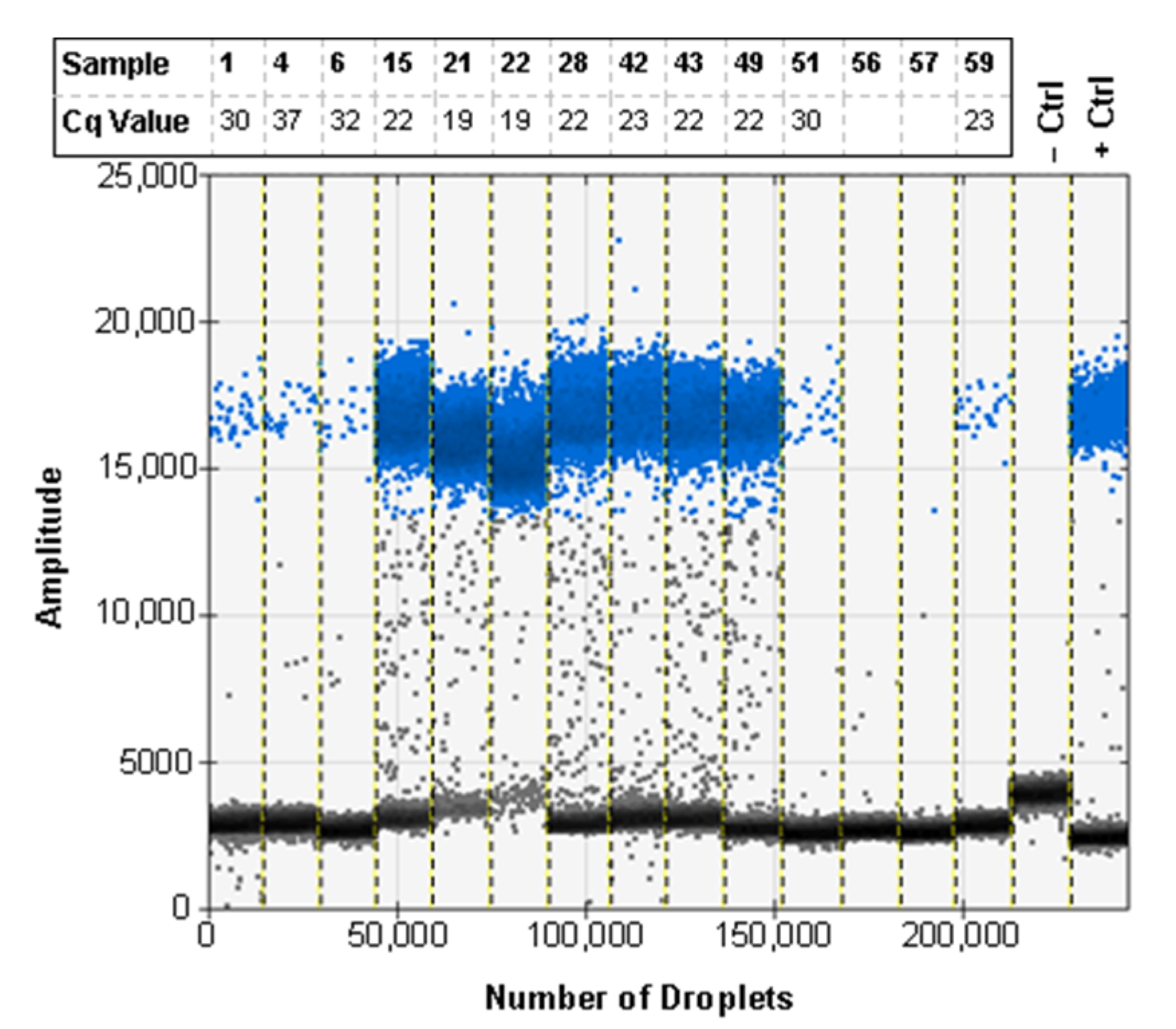
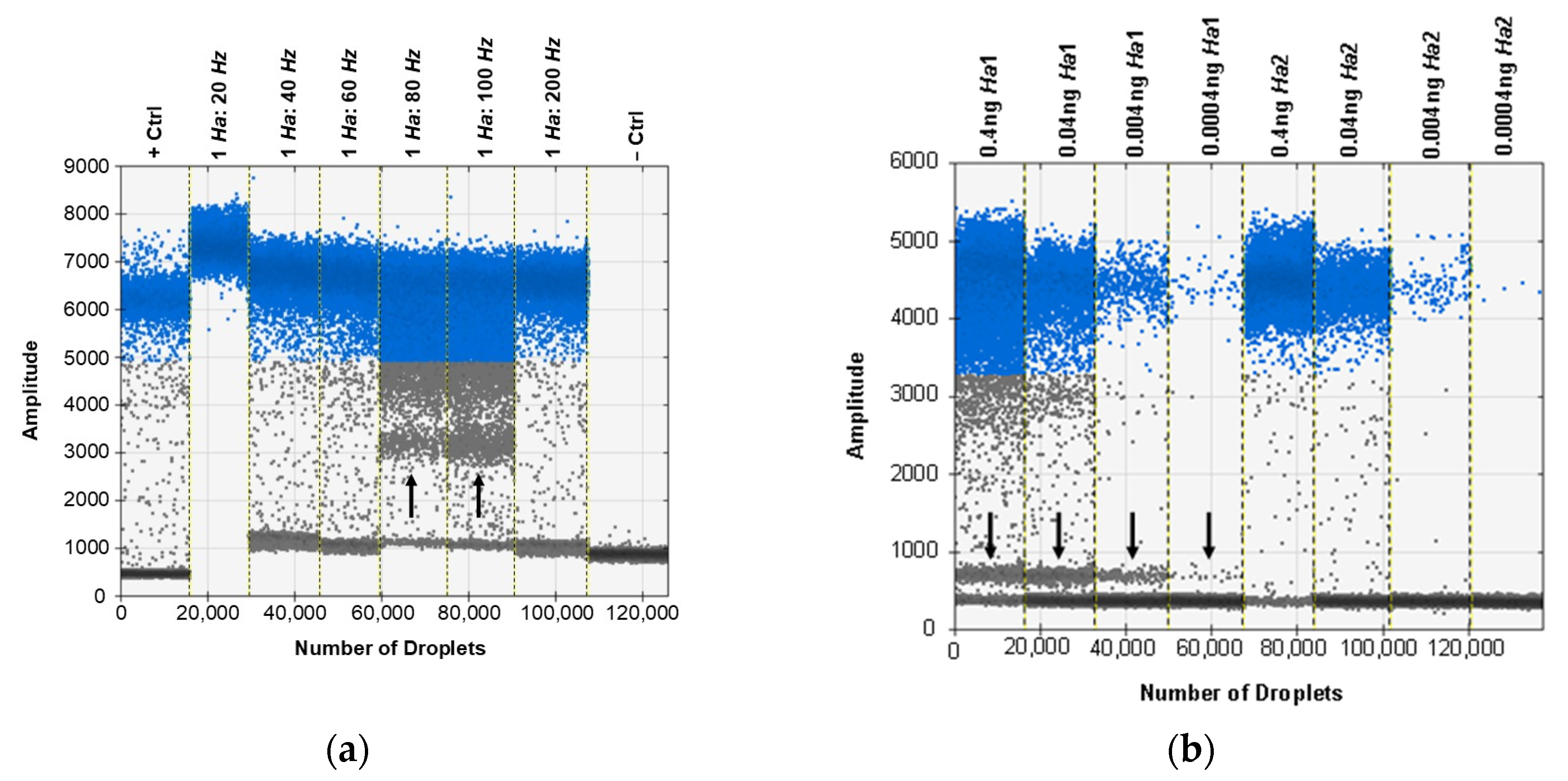
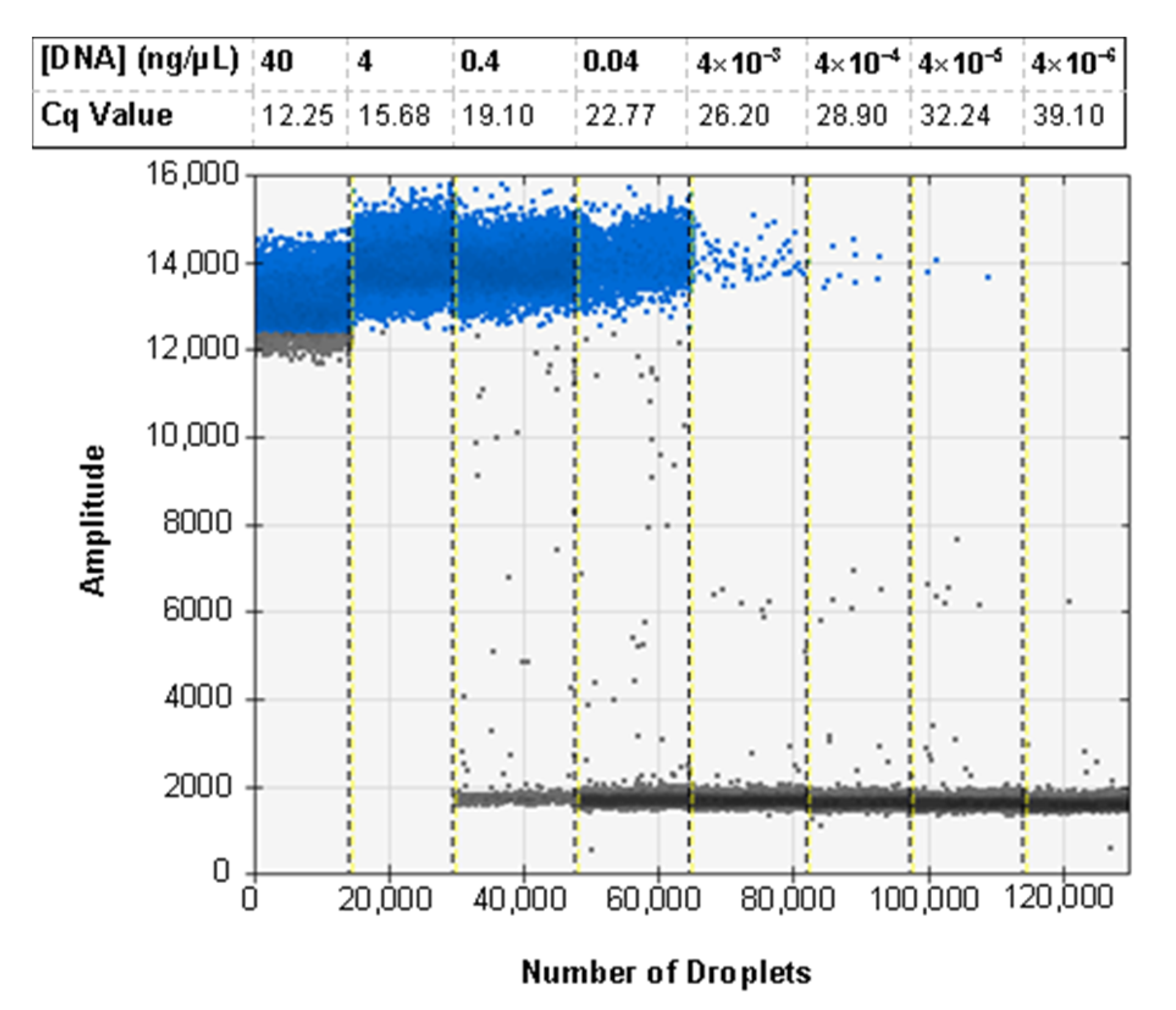
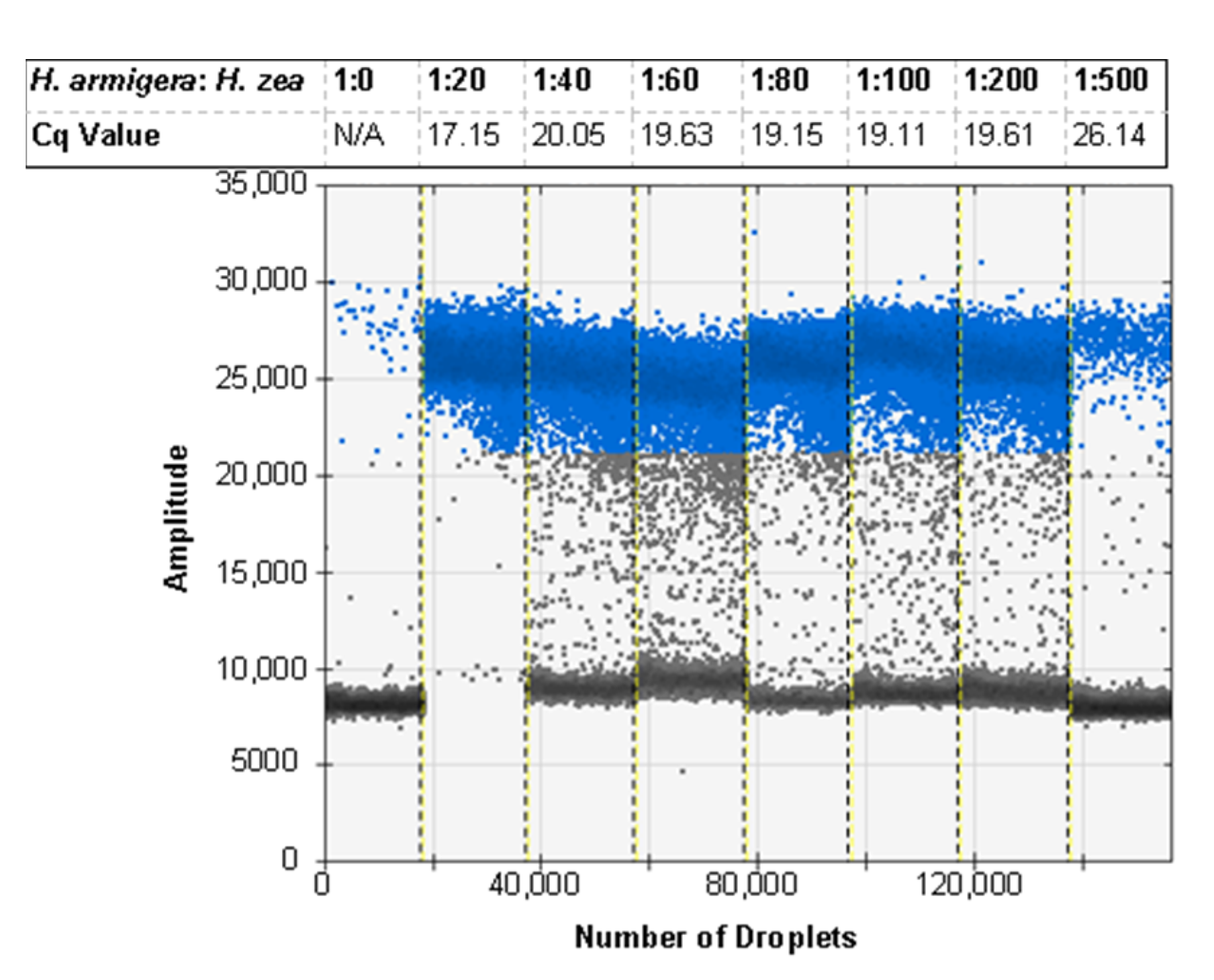
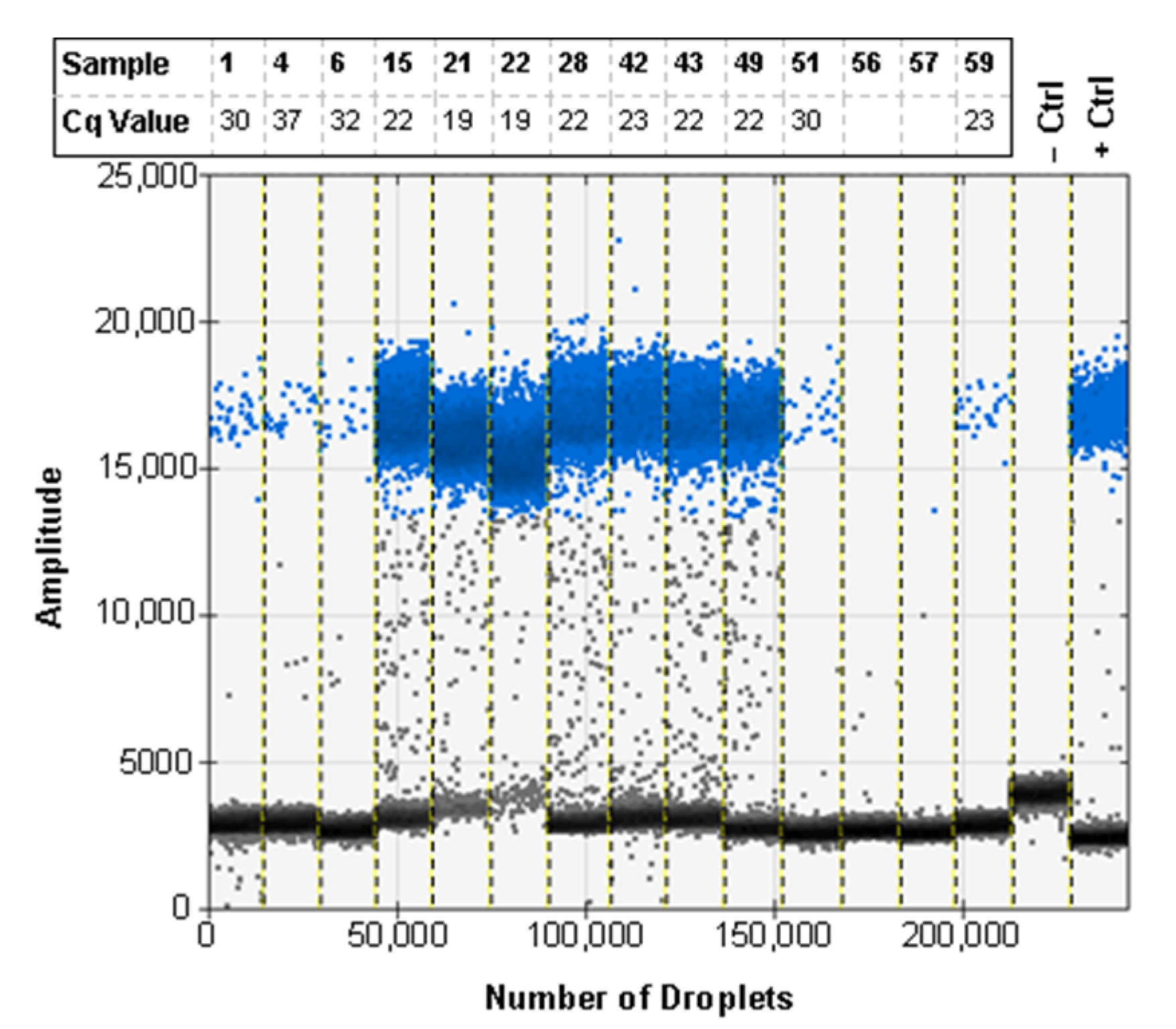
3.6. ddPCR Bulk Sample Results and Relative Performance of EvaGreen and Probe-Based Assays
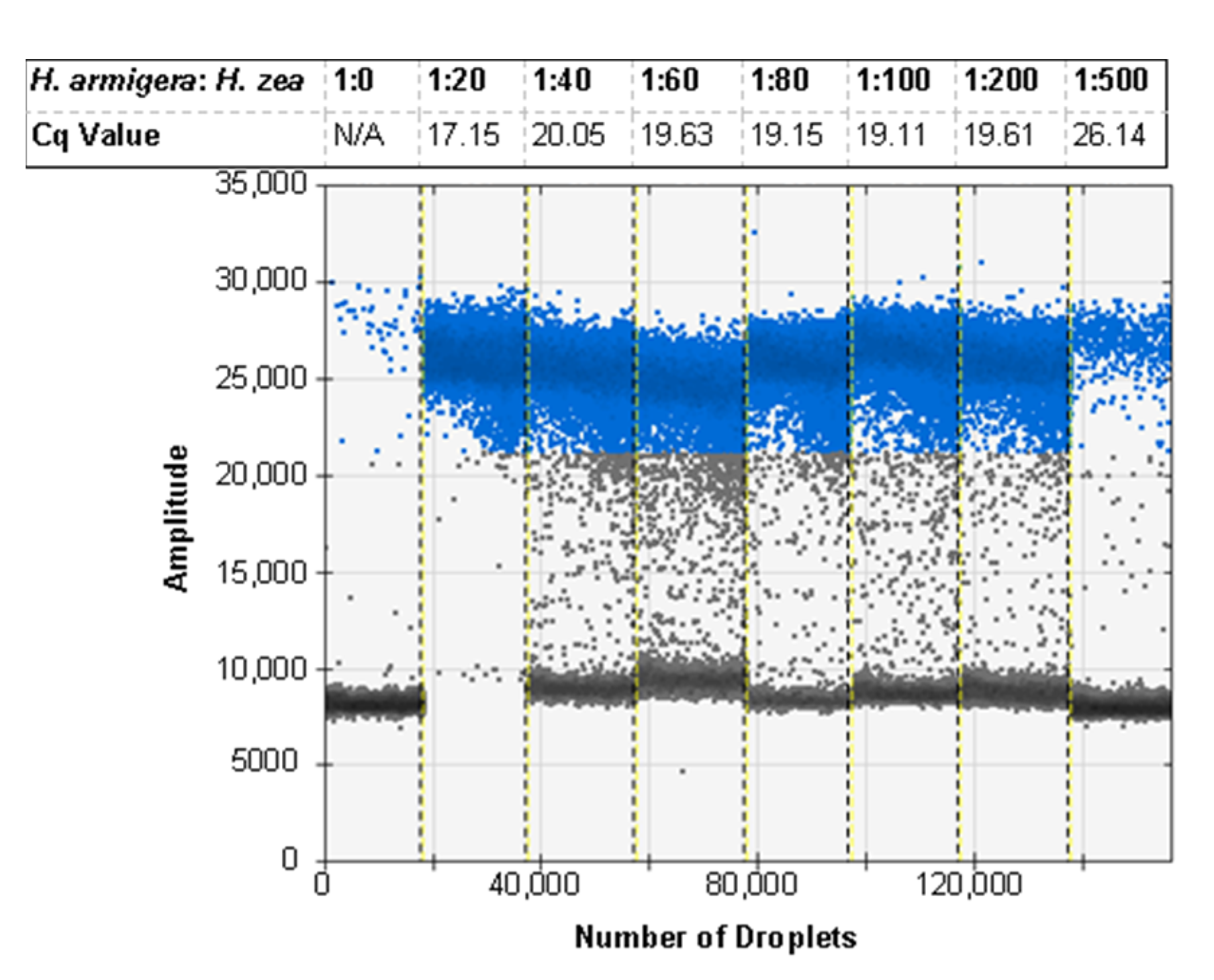
3.7. ddPCR False Positive Rate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kogan, M.; Helm, C.G.; Kogan, J.; Brewer, E. Distribution and economic importance of Heliothis virescens and Heliothis zea in North, Central, and South America and of their natural enemies and host plants. In Proceedings of the Workshop on Biological Control of Heliothis: Increasing the Effectiveness of Natural Enemies, Illustrated ed.; King, E.G., Jackson, R.D., Eds.; Far Eastern Regional Research Services, Office of International Cooperation and Development, US Department of Agriculture: New Delhi, India, 1989; p. 550. [Google Scholar]
- Hardwick, D.F. A Monograph of the North American Heliothentinae (Lepidoptera: Noctuidae); D. F. Hardwick: Ottawa, ON, Canada, 1996; p. 281. [Google Scholar]
- Blanco, C.A.; Terán-Vargas, A.P.; López, J.D., Jr.; Kauffman, J.V.; Wei, X.-K. Densities of Heliothis virescens and Helicoverpa zea (Lepidoptera: Noctuidae) in three plant hosts. Fla. Entomol. 2007, 90, 742–750. [Google Scholar] [CrossRef]
- Cunningham, J.P.; Zalucki, M.P. Understanding heliothine (Lepidoptera: Heliothinae) pests: What is a host plant? J. Econ. Entomol. 2014, 107, 881–896. [Google Scholar] [CrossRef]
- Cezpak, C.; Albernaz, K.C.; Vivan, L.M.; Guimarães, H.O.; Carvalhais, T. First reported occurrence of Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae) in Brazil. Presquisa Agropecuária Trop. 2013, 43, 110–113. [Google Scholar]
- Tay, W.T.; Soria, M.F.; Walsh, T.; Thomazoni, D.; Silvie, P.; Behere, G.T.; Anderson, C.; Downes, S. A brave new world for an old world pest: Helicoverpa armigera (Lepidoptera: Noctuidae) in Brazil. PLoS ONE 2013, 8, e80134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardwick, D.F. The corn earworm complex. Mem. Entomol. Soc. Can. 1965, 97, 5–247. [Google Scholar] [CrossRef]
- Jones, C.M.; Parry, H.; Tay, W.T.; Reynolds, D.R.; Chapman, J.W. Movement Ecology of Pest Helicoverpa: Implications for Ongoing Spread. Annu. Rev. Entomol. 2019, 64, 277–295. [Google Scholar] [CrossRef] [Green Version]
- Smith, E. Detection of old world bollworm (Helicoverpa armigera) in Puerto Rico. 2014. [Google Scholar]
- Hayden, J.; Brambila, J. Pest alert: Helicoverpa armigera (Lepidoptera: Noctuidae), the Old World Bollworm. 2015. [Google Scholar]
- Jones, C.M.; Papanicolaou, A.; Mironidis, G.K.; Vontas, J.; Yang, Y.; Lim, K.S.; Oakeshott, J.G.; Bass, C.; Chapman, J.W. Genomewide transcriptional signatures of migratory flight activity in a globally invasive insect pest. Mol. Ecol. 2015, 24, 4901–4911. [Google Scholar] [CrossRef] [Green Version]
- Tay, W.T.; Walsh, T.K.; Downes, S.; Anderson, C.; Jermiin, L.S.; Wong, T.K.; Piper, M.C.; Chang, E.S.; Macedo, I.B.; Czepak, C.; et al. Mitochondrial DNA and trade data support multiple origins of Helicoverpa armigera (Lepidoptera, Noctuidae) in Brazil. Sci. Rep. 2017, 7, 45302. [Google Scholar] [CrossRef] [Green Version]
- Tembrock, L.R.; Timm, A.E.; Zink, F.A.; Gilligan, T.M. Phylogeography of the Recent Expansion of Helicoverpa armigera (Lepidoptera: Noctuidae) in South America and the Caribbean Basin. Ann. Entomol. Soc. Am. 2019, 112, 388–401. [Google Scholar] [CrossRef] [Green Version]
- Gilligan, T.M.; Goldstein, P.Z.; Timm, A.E.; Farris, R.; Ledezma, L.; Cunningham, A.P. Identification of Heliothine (Lepidoptera: Noctuidae) Larvae Intercepted at US Ports of Entry From the New World. J. Econ. Entomol. 2019, 112, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Fowler, G.; Lakin, K. Risk Assessment: The Old World Bollworm, Helicoverpa armigera (Hübner) (Leipdoptera: Noctuidae). 2001; 1–19. [Google Scholar]
- Kriticos, D.J.; Ota, N.; Hutchison, W.D.; Beddow, J.; Walsh, T.; Tay, W.T.; Borchert, D.M.; Paula-Moreas, S.V.; Czepak, C.; Zalucki, M.P. The potential distribution of invading Helicoverpa armigera in North America: Is it just a matter of time? PLoS ONE 2015, 10, e0119618. [Google Scholar] [CrossRef] [Green Version]
- Haile, F.; Nowatzki, T.; Storer, N.; Davis, J. Overview of Pest Status, Potential Risk, and Management Considerations of Helicoverpa armigera (Lepidoptera: Noctuidae) for U.S. Soybean Production. J. Integr. Pest Manag. 2021, 12, 3. [Google Scholar] [CrossRef]
- Pogue, M.G. A new synonym of Helicoverpa zea (Bodie) and differentiation of adult males of H. zea and H. armigera (Hubner) (Lepidoptera: Noctuidae: Heliothinae). Ann. Entomol. Soc. Am. 2004, 97, 1222–1226. [Google Scholar] [CrossRef] [Green Version]
- Brambila, J. Instructions for dissecting male genitalia of Helicoverpa (Lepidoptera: Noctuidae) to separate H. zea from H. armigera (Hubner). 2009. [Google Scholar]
- Anderson, C.J.; Oakeshott, J.G.; Tay, W.T.; Gordon, K.H.J.; Zwick, A.; Walsh, T.K. Hybridization and gene flow in the mega-pest lineage of moth, Helicoverpa. Proc. Natl. Acad. Sci. USA 2018, 115, 5034–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Bai, R.; Zhao, Z.; Tao, L.; Ma, M.; Ji, Z.; Jian, M.; Ding, Z.; Dai, X.; Bao, F.; et al. Application of droplet digital PCR to detect the pathogens of infectious diseases. Biosci. Rep. 2018, 38, BSR20181170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zink, F.A.; Tembrock, L.R.; Timm, A.E.; Gilligan, T.M. A duplex ddPCR assay for simultaneously detecting Ips sexdentatus and Ips typographus (Coleoptera: Curculionidae) in bulk trap samples. Can. J. For. Res. 2019, 49, 903–914. [Google Scholar] [CrossRef]
- Capo, E.; Spong, G.; Koizumi, S.; Puts, I.; Olajas, F.; Königsson, H.; Karlsson, J.; By-ström, P. Droplet digital PCR applied to environmental DNA, a promising method to estimate fish population abundance from humic-rich aquatic ecosystems. Environ. DNA 2021, 3, 343–352. [Google Scholar] [CrossRef]
- Zink, F.A.; Tembrock, L.R.; Timm, A.E.; Farris, R.E.; Perera, O.P.; Gilligan, T.M. A droplet digital PCR (ddPCR) assay to detect Helicoverpa armigera (Lepidoptera: Noctuidae) in bulk trap samples. PLoS ONE 2017, 12, e0178704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M. Digital PCR hits its stride. Nat. Methods 2012, 9, 541–544. [Google Scholar] [CrossRef]
- Solà, M.; Lundgren, J.G.; Agustí, N.; Riudavets, J. Detection and quantification of the isect pest Rhyzopertha dominica (F.) (Coleoptera: Bostrichidae) in rice by qPCR. J. Stored Prod. Res. 2017, 71, 106–111. [Google Scholar] [CrossRef]
- Tembrock, L.R.; Farris, R.E.; Ledezma, L.; Barr, N.B.; Gilligan, T.M. A Real-Time PCR Assay for the Separation of Autographa gamma (Noctuidae: Plusiinae) From Morphologically Similar Species in North America. J. Econ. Entomol. 2017, 110, 2609–2617. [Google Scholar] [CrossRef]
- Capron, A.; Stewart, D.; Hrywkiw, K.; Allen, K.; Feau, N.; Bilodeau, G.; Tanguay, P.; Cusson, M.; Hamelin, R.C. In situ processing and efficient environmental detection (iSPEED) of tree pests and pathogens using point-of-use real-time PCR. PLoS ONE 2020, 15, e0226863. [Google Scholar] [CrossRef]
- Perera, O.P.; Allen, K.C.; Jain, D.; Purcell, M.; Little, N.S.; Luttrell, R.G. Rapid identification of Helicoverpa armigera and Helicoverpa zea (Lepidoptera: Noctuidae) using ribosomal RNA internal transcribed spacer 1. J. Insect Sci. 2015, 15, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloor, G.B.; Preston, C.R.; Johnson-Schlitz, D.M.; Nassif, N.A.; Phillis, R.W.; Benz, W.K.; Robertson, H.M.; Engels, W.R. Type I repressors of P element mobility. Genetics 1993, 135, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, T.M.; Tembrock, L.R.; Farris, R.E.; Barr, N.B.; van der Straten, M.J.; van de Vossenberg, B.T.; Metz-Verschure, E. A Multiplex Real-Time PCR Assay to Diagnose and Separate Helicoverpa armigera and H. zea (Lepidoptera: Noctuidae) in the New World. PLoS ONE 2015, 10, e0142912. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SantaLucia, J.J. A unified view of polymer, dumbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef] [PubMed]
- Barr, N.B.; Ledezma, L.A.; Farris, R.E.; Epsteirn, M.E.; Gilligan, T.M. A multiplex real-time polymerase chin reaction assay to diagnose Epiphyas postvittana (Lepidoptera: Tortricidae). J. Econ. Entomol. 2011, 104, 1706–1719. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statisticl Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Grothendieck, G. nls2: Non-Linear Regression with Brute Force, R package version 2.0. 2013.
- Mendiburu, F. Agricolae: Statistical Procedures from Agricultural Research, 1.3-3, R. 2020.
- Zink, F.A.; Tembrock, L.R.; Timm, A.E.; Gilligan, T.M. A ddPCR Assay for Identification of Autographa gamma (Noctuidae: Plusiinae) in Bulk Trap Samples. J. Econ. Entomol. 2018, 111, 1490–1495. [Google Scholar] [CrossRef]
- Jones, M.; Williams, J.; Gartner, K.; Phillips, R.; Hurst, J.; Frater, J. Low copy target detection by Droplet Digital PCR through application of a novel open access bioinformatic pipeline, ‘definetherain’. J. Virol. Methods 2014, 202, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. 1), S49–S52. [Google Scholar] [PubMed]
- Walsh, P.S.; Erlich, H.A.; Higuchi, R. Preferential PCR amplification of alleles: Mechanisms and solutions. PCR Methods Appl. 1992, 1, 241–250. [Google Scholar] [CrossRef]
- Elnifro, E.M.; Ashshi, A.M.; Cooper, R.J.; Klapper, P.E. Multiplex PCR: Optimization and application in diagnostic virology. Clin. Microbiol. Rev. 2000, 13, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Sint, D.; Raso, L.; Traugott, M. Advances in multiplex PCR: Balancing primer efficiencies and improving detection success. Methods Ecol. Evol. 2012, 3, 898–905. [Google Scholar] [CrossRef]
- Quan, P.L.; Sauzade, M.; Brouzes, E. dPCR: A Technology Review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef] [Green Version]
- Mahendran, P.; Liew, J.W.K.; Amir, A.; Ching, X.T.; Lau, Y.L. Droplet digital polymerase chain reaction (ddPCR) for the detection of Plasmodium knowlesi and Plasmodium vivax. Malar. J. 2020, 19, 241. [Google Scholar] [CrossRef]
- Prokopowich, C.D.; Gregory, T.R.; Crease, T.J. The correlation between rDNA copy number and genome size in eukaryotes. Genome 2003, 46, 48–50. [Google Scholar] [CrossRef]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Tembrock, L.R.; Zink, F.A.; Timm, A.E.; Gilligan, T.M. A HiRise Genome for Helicoverpa zea × H. armigera hybrids. 2021; manuscript in preparation. [Google Scholar]
- Nei, M.; Rooney, A.P. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 2005, 39, 121–152. [Google Scholar] [CrossRef] [Green Version]
- Freire, R.; Arias, A.; Insua, A.M.; Mendez, J.; Eirin-Lopez, J.M. Evolutionary dynamics of the 5S rDNA gene family in the mussel Mytilus: Mixed effects of birth-and-death and concerted evolution. J. Mol. Evol. 2010, 70, 413–426. [Google Scholar] [CrossRef]
- Teruel, M.; Ruíz-Ruano, F.J.; Marchal, J.A.; Sánchez, A.; Cabrero, J.; Camacho, J.P.; Perfectti, F. Disparate molecular evolution of two types of repetitive DNAs in teh genome of the grasshopper Eyprepocnemis plorans. Heredity 2014, 112, 531–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dMIQE Group; Huggett, J.F. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.S.; Pienaar, E.; Termaat, J.R.; Whitney, S.E.; Louw, T.M.; Viljoen, H.J. Efficiency of the Polymerase Chain Reaction. Chem. Eng. Sci. 2010, 65, 4996–5006. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Sequence | Tm (°C) | Source |
|---|---|---|---|---|
| Harm_18S_1944F | Diagnostic | 5′-AACGTAAACAATAATCCACACACCA | 55.1 | This study |
| Harm_18S_2154R | Diagnostic | 5′-CGCGGATTTTTGTGTTTTGTGT | 56.5 | This study |
| Harm_18S_1969P | Diagnostic | 5′-6-FAM-CTAGAGGAC-ZEN-ACAGAGTCGAACG-IowaBlackFQ | 56.4 | This study |
| RT-18S-F2 | Control | 5′-ACCGCCCTAGTTCTAACCGTAAA | 57.8 | [35] |
| RT-18S-R2 | Control | 5′-CCGCCGAGCCATTGTAGTAA | 57.3 | [35] |
| RT-18S-P2 | Control | 5′-Quasar670-TGTCATCTAGCGATCCGCCGA-BHQ-2 | 60.3 | [35] |
| rcf (xg) | NaCl Conc. (mM) | EDTA Conc. (mM) | Cq |
|---|---|---|---|
| 2152 | 12.5 | 0.5 | 23.33 |
| 16,873 | 12.5 | 0.5 | 20.39 |
| 2152 | 25 | 1 | 21.96 |
| 16,873 | 25 | 1 | 19.16 |
| 2152 | 50 | 2 | 18.92 |
| 16,863 | 50 | 2 | 18.33 |
| Leg Ratio | rcf (xg) | NaCl Conc. (mM) | EDTA Conc. (mM) | Cq |
|---|---|---|---|---|
| 1:20 | 2152 | 50 | 2 | 19.00 |
| 1:20 | 16,873 | 50 | 2 | 18.29 |
| 1:20 | 2152 | 250 | 10 | 20.58 |
| 1:20 | 16,873 | 250 | 10 | 18.15 |
| 1:20 | 8609 | 100 | 5 | 17.16 |
| 1:60 | 8609 | 100 | 5 | 20.36 |
| Ratio | FAM Cq |
|---|---|
| 1:20 | 17.15 |
| 1:40 | 20.05 |
| 1:60 | 19.63 |
| 1:80 | 19.15 |
| 1:100 | 19.11 |
| 1:200 | 19.61 |
| 1:500 | 26.14 |
| 1:0 | 18.44 |
| 0:1 | |
| NTC |
| −Ctrl/−BP | −Ctrl/+BP | +Ctrl/−BP | +Ctrl/+BP | |||||
|---|---|---|---|---|---|---|---|---|
| Sample | Cq | RFU | Cq | RFU | Cq | RFU | Cq | RFU |
| 1 | 31.65 | 198.94 | 30.13 | 2291.84 | 11.58 | 30.11 | 402.18 | |
| 2 | 17.11 | 31.32 | 573.64 | 14.32 | 35.72 | 116.86 | ||
| 3 | 27.58 | 1208.05 | 28.10 | 4182.44 | 30.20 | 242.66 | 27.36 | 1547.18 |
| 4 | 10.99 | 34.51 | 216.99 | 14.23 | 36.53 | 107.33 | ||
| 5 | 25.07 | 3302.44 | 25.69 | 7287.27 | 25.36 | 980.38 | 26.04 | 1531.11 |
| 6 | 8.74 | 35.72 | 127.97 | 20.37 | 32.15 | 263.40 | ||
| 7 | 22.18 | 8860.18 | 22.43 | 12,029.25 | 21.59 | 1303.99 | 21.76 | 10,240.34 |
| 8 | 28.67 | 221.20 | 26.02 | 5089.04 | 22.56 | 25.97 | 2504.50 | |
| 9 | 20.53 | 14,640.72 | 20.91 | 11,983.67 | 20.18 | 8987.33 | 21.09 | 4915.74 |
| 10 | 20.60 | 12,828.26 | 21.63 | 11,260.27 | 21.32 | 8552.34 | 21.87 | 7851.89 |
| 11 | 20.69 | 6733.72 | 21.12 | 14,552.49 | 20.78 | 5623.11 | 21.11 | 9787.14 |
| 12 | 26.21 | 2500.98 | 25.95 | 8619.49 | 25.51 | 1333.16 | 25.81 | 3735.53 |
| 13 | 26.69 | 7189.02 | 27.17 | 6005.49 | 25.14 | 591.95 | 27.02 | 5404.36 |
| 14 | 27.95 | 2879.51 | 26.02 | 9604.84 | 27.38 | 465.66 | 25.38 | 3628.56 |
| 15 | 19.83 | 12,057.90 | 21.83 | 12,285.58 | 20.30 | 9984.92 | 22.09 | 9215.15 |
| 16 | 20.50 | 14,147.07 | 21.46 | 12,672.84 | 20.44 | 7220.47 | 21.42 | 10,179.95 |
| 17 | 19.45 | 10,313.26 | 21.49 | 13,419.63 | 20.47 | 6204.70 | 21.32 | 10,309.40 |
| 18 | 19.73 | 10,536.28 | 21.11 | 13,353.52 | 20.62 | 9439.42 | 21.27 | 10,526.17 |
| 19 | 21.17 | 14,933.12 | 21.82 | 15,438.62 | 18.63 | 4943.59 | 21.12 | 12,605.58 |
| 20 | 19.28 | 14,301.02 | 21.78 | 15,255.25 | 19.73 | 8265.60 | 23.67 | 8443.85 |
| 21 | 19.60 | 7794.62 | 20.00 | 16,928.45 | 18.93 | 9460.50 | 19.16 | 13,813.90 |
| 22 | 19.18 | 9441.36 | 19.20 | 17,116.53 | 18.64 | 3999.75 | 19.09 | 14,047.80 |
| 23 | 21.21 | 10,290.38 | 21.31 | 14,504.58 | 21.43 | 11,550.59 | 21.94 | 11,009.13 |
| 24 | 29.23 | 229.14 | 27.10 | 6704.37 | 27.29 | 460.06 | 26.87 | 1772.06 |
| 25 | 25.03 | 3004.36 | 25.06 | 10,938.61 | 25.22 | 915.49 | 24.39 | 5906.28 |
| 26 | 21.25 | 9820.98 | 21.48 | 11,949.11 | 21.28 | 5401.31 | 21.24 | 11,309.96 |
| 27 | 20.37 | 11,952.59 | 19.72 | 15,220.42 | 20.37 | 9794.59 | 19.88 | 12,599.93 |
| 28 | 22.27 | 12,402.87 | 22.79 | 11,506.99 | 22.06 | 4630.17 | 22.07 | 9768.89 |
| 29 | 23.47 | 10,129.45 | 22.37 | 4684.52 | 23.39 | 3080.91 | 23.34 | 8869.53 |
| 30 | 21.65 | 9775.21 | 22.61 | 13,290.02 | 21.55 | 7357.58 | 22.21 | 6413.43 |
| 31 | 25.66 | 2216.63 | 22.72 | 6769.11 | 25.47 | 765.97 | 23.04 | 5099.04 |
| 32 | 20.23 | 11,324.96 | 20.26 | 16,288.94 | 19.95 | 7612.89 | 20.16 | 11,456.42 |
| 33 | 20.87 | 5143.82 | 19.55 | 16,991.30 | 23.07 | 19.43 | 11,137.10 | |
| 34 | 20.25 | 13,967.67 | 20.69 | 15,999.46 | 20.90 | 11,097.20 | 20.39 | 12,280.23 |
| 35 | 22.06 | 11,644.17 | 21.87 | 15,842.31 | 21.53 | 3371.78 | 22.43 | 10,471.81 |
| 36 | 28.31 | 3564.74 | 28.53 | 2762.28 | 28.75 | 267.93 | 28.41 | 1097.53 |
| 37 | 22.12 | 6009.29 | 21.95 | 15,605.20 | 23.25 | 519.00 | 21.59 | 10,985.13 |
| 38 | 19.33 | 14,362.07 | 21.14 | 16,877.70 | 21.63 | 346.42 | 20.31 | 12,842.48 |
| 39 | 22.20 | 11,286.24 | 22.13 | 14,702.44 | 21.37 | 4716.22 | 21.38 | 10,587.83 |
| 40 | 23.65 | 8505.87 | 24.17 | 10,657.79 | 23.44 | 4196.02 | 23.69 | 8107.57 |
| 41 | 28.61 | 377.28 | 27.06 | 6554.71 | 33.05 | 140.50 | 26.21 | 2651.93 |
| 42 | 23.43 | 7592.74 | 23.35 | 12,718.12 | 17.83 | 22.75 | 9158.52 | |
| 43 | 22.34 | 4626.78 | 22.43 | 11,485.15 | 28.37 | 252.15 | 22.42 | 9117.53 |
| 44 | 20.55 | 8772.13 | 20.12 | 17,240.06 | 21.34 | 1286.78 | 20.51 | 11,953.43 |
| 45 | 20.43 | 10,108.94 | 19.98 | 12,116.37 | 21.58 | 1636.07 | 20.40 | 10,375.38 |
| 46 | 25.23 | 2868.87 | 24.40 | 11,150.40 | 25.01 | 2595.12 | 24.80 | 3996.54 |
| 47 | 22.43 | 2316.19 | 22.52 | 7676.01 | 22.47 | 6662.04 | 23.15 | 6540.77 |
| 48 | 5.68 | 26.96 | 6957.75 | 10.08 | 27.34 | 508.59 | ||
| 49 | 22.12 | 5735.89 | 23.10 | 13,885.75 | 21.76 | 4549.79 | 22.07 | 10,361.06 |
| 50 | 30.12 | 185.46 | 27.07 | 4677.50 | 34.21 | 120.88 | 25.79 | 1623.45 |
| 51 | 29.38 | 1970.22 | 29.74 | 1305.81 | 37.49 | 29.96 | 977.40 | |
| 52 | 26.19 | 7288.93 | 25.82 | 7727.24 | 25.63 | 2584.78 | 25.35 | 6046.17 |
| 53 | 26.95 | 4197.06 | 27.62 | 5171.64 | 27.50 | 298.00 | 27.43 | 864.95 |
| 54 | 24.32 | 9713.18 | 25.29 | 8753.72 | 24.69 | 2931.30 | 25.21 | 3978.88 |
| 55 | 22.44 | 11,339.61 | 23.09 | 3627.23 | 22.84 | 3692.81 | 23.28 | 5743.53 |
| 56 | 6.94 | 32.23 | 362.75 | 6.86 | 84.44 | |||
| 57 | 15.72 | 31.32 | 544.89 | 9.25 | 88.04 | |||
| 58 | 39.24 | 90.50 1 | 31.40 | 582.00 | 8.51 | 34.41 | 141.23 | |
| 59 | 22.31 | 9172.41 | 22.08 | 9900.71 | 22.07 | 5431.74 | 22.83 | 8343.40 |
| Variation Source | df | Sum Square | Mean Square | F | p |
|---|---|---|---|---|---|
| Bulk sample | 58 | 3,952,285,666 | 68,142,856 | 13.018 | <2 × 10−16 *** |
| Use of 18S control (Ctrl) | 1 | 546,435,572 | 546,435,572 | 104.39 | <2 × 10−16 *** |
| Use of bead purification (BP) | 1 | 561,294,724 | 561,294,724 | 107.23 | <2 × 10−16 *** |
| Ctrl × BP | 1 | 545,735 | 545,735 | 0.1043 | 0.7472 |
| Residuals | 171 | 895,103,428 | 5,234,523 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, T.M.R.; Zink, F.A.; Menezes, R.C.; Dianese, É.C.; Albernaz-Godinho, K.C.; Cunha, M.G.; Timm, A.E.; Gilligan, T.M.; Tembrock, L.R. Assay Optimization Can Equalize the Sensitivity of Real-Time PCR with ddPCR for Detection of Helicoverpa armigera (Lepidoptera: Noctuidae) in Bulk Samples. Insects 2021, 12, 885. https://doi.org/10.3390/insects12100885
Oliveira TMR, Zink FA, Menezes RC, Dianese ÉC, Albernaz-Godinho KC, Cunha MG, Timm AE, Gilligan TM, Tembrock LR. Assay Optimization Can Equalize the Sensitivity of Real-Time PCR with ddPCR for Detection of Helicoverpa armigera (Lepidoptera: Noctuidae) in Bulk Samples. Insects. 2021; 12(10):885. https://doi.org/10.3390/insects12100885
Chicago/Turabian StyleOliveira, Thayssa M. R., Frida A. Zink, Renato C. Menezes, Érico C. Dianese, Karina C. Albernaz-Godinho, Marcos G. Cunha, Alicia E. Timm, Todd M. Gilligan, and Luke R. Tembrock. 2021. "Assay Optimization Can Equalize the Sensitivity of Real-Time PCR with ddPCR for Detection of Helicoverpa armigera (Lepidoptera: Noctuidae) in Bulk Samples" Insects 12, no. 10: 885. https://doi.org/10.3390/insects12100885
APA StyleOliveira, T. M. R., Zink, F. A., Menezes, R. C., Dianese, É. C., Albernaz-Godinho, K. C., Cunha, M. G., Timm, A. E., Gilligan, T. M., & Tembrock, L. R. (2021). Assay Optimization Can Equalize the Sensitivity of Real-Time PCR with ddPCR for Detection of Helicoverpa armigera (Lepidoptera: Noctuidae) in Bulk Samples. Insects, 12(10), 885. https://doi.org/10.3390/insects12100885