An Alternative, High Throughput Method to Identify Csd Alleles of the Honey Bee

 and
and
Abstract
1. Introduction
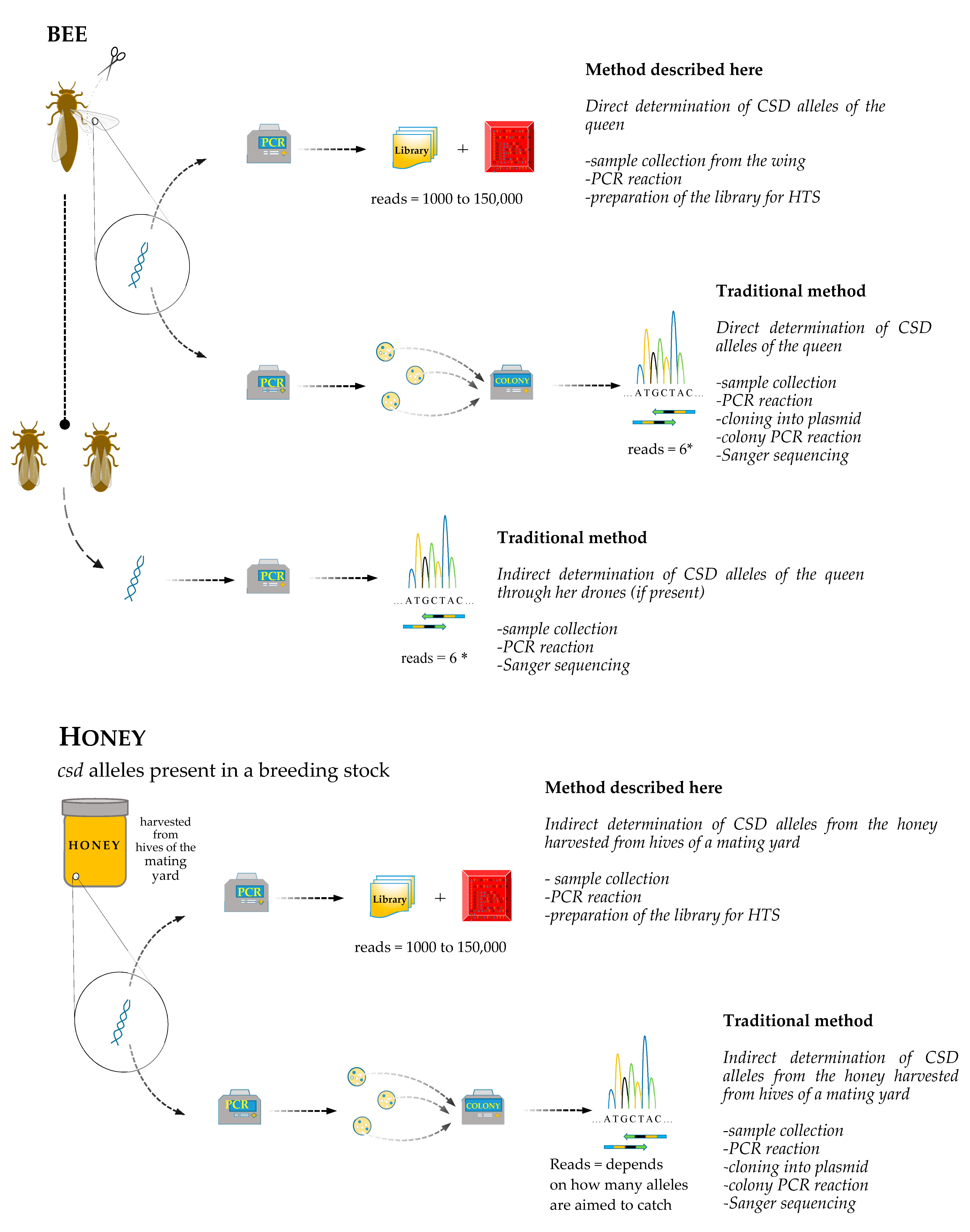
2. Material and Methods
2.1. DNA Extraction from Bees
2.2. DNA Extraction from Honey
2.3. Sequencing of csd Alleles
2.4. Bioinformatic Analysis
3. Results
3.1. Individual csd Alleles Might Be Genotyped from Samples Containing Multiple Alleles
3.2. Novel csd Alleles Were Uncovered Using High Throughput Sequencing
4. Discussion
4.1. A High-Throughput Sequencing Technique is Provided for Detection of the csd Alleles of a Breeding Queen
4.2. High Throughput Determination of csd Alleles from Honey Might Open the Way for Large-Scale Screening of Breeding Stocks But Has Limitations
4.3. Tracking of csd Alleles Offers Potential Benefits for Closed Breeding Programmes Based on Instrumental Insemination
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilson, E.B. The chromosomes in relation to the determination of sex in insects. Science 1905, 22, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Dzierzon, J. Gutachten über die von Herrn Direktor Stöhr im ersten und zweiten Kapitel des General-Gutachtens aufgestellten Fragen. Eichstädter Bienenztg. 1845, 1, 119–121. [Google Scholar]
- Bull, J.J. Evolution of Sex Determining Mechanisms; The Benjamin/Cummings Publishing Company, Inc.: San Francisco, CA, USA, 1983. [Google Scholar]
- Cook, J.M. Sex determination in the Hymenoptera: A review of models and evidence. Heredity 1993, 71, 421–435. [Google Scholar] [CrossRef]
- Whiting, P.; Whiting, A.R. Diploid males from fertilized eggs in Hymenoptera. Science 1925, 62, 437. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, O. Viability and sex determination in the honey bee (Apis mellifera L.). Genetics 1951, 36, 500–509. [Google Scholar]
- Kaskinova, M.; Nikolenko, A. csd gene of honeybee: Genetic structure, functioning, and evolution. Russian J. Gen. 2017, 53, 297–301. [Google Scholar] [CrossRef]
- Beye, M.; Seelmann, C.; Gempe, T.; Hasselmann, M.; Vekemans, X.; Fondrk, M.K.; Page, R.E., Jr. Gradual molecular evolution of a sex determination switch through incomplete penetrance of femaleness. Curr. Biol. 2013, 23, 2559–2564. [Google Scholar] [CrossRef]
- Beye, M.; Hasselmann, M.; Fondrk, M.K.; Page, R.E., Jr.; Omholt, S.W. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell 2003, 114, 419–429. [Google Scholar] [CrossRef]
- Harpur, B.A.; Sobhani, M.; Zayed, A. A review of the consequences of complementary sex determination and diploid male production on mating failures in the Hymenoptera. Entomol. Exp. Appl. 2013, 146, 156–164. [Google Scholar] [CrossRef]
- Hyink, O.; Laas, F.; Dearden, P.K. Genetic tests for alleles of complementary-sex-determiner to support honeybee breeding programmes. Apidologie 2013, 44, 306–313. [Google Scholar] [CrossRef]
- Franck, P.; Garnery, L.; Solignac, M.; Cornuet, J.M. Molecular confirmation of a fourth lineage in honeybees from the Near East. Apidologie 2000, 31, 167–180. [Google Scholar] [CrossRef]
- Ruttner, F. Morphometric Analysis and Classification, in Biogeography and Taxonomy of Honeybees; Springer: Berlin/Heidelberg, Germany, 1988; pp. 66–78. [Google Scholar]
- Ruttner, F.; Tassencourt, L.; Louveaux, J. Biometrical-statistical analysis of the geographic variability of Apis mellifera LI Material and methods. Apidologie 1978, 9, 363–381. [Google Scholar] [CrossRef]
- Moritz, R.F.; Härtel, S.; Neumann, P. Global invasions of the western honeybee (Apis mellifera) and the consequences for biodiversity. Ecoscience 2005, 12, 289–301. [Google Scholar] [CrossRef]
- Cho, S.; Huang, Z.Y.; Green, D.R.; Smith, D.R.; Zhang, J. Evolution of the complementary sex-determination gene of honey bees: Balancing selection and trans-species polymorphisms. Genome Res. 2006, 16, 1366–1375. [Google Scholar] [CrossRef]
- Kaskinova, M.D.; Gataullin, A.R.; Saltykova, E.S.; Gaifullina, L.R.; Poskryakov, A.V.; Nikolenko, A.G. Polymorphism of the Hypervariable Region of the csd Gene in the Apis mellifera L. Population in Southern Urals. Russian J. Gen. 2019, 55, 267–270. [Google Scholar] [CrossRef]
- Zareba, J.; Blazej, P.; Laszkiewicz, A.; Sniezewski, L.; Majkowski, M.; Janik, S.; Cebrat, M. Uneven distribution of complementary sex determiner (csd) alleles in Apis mellifera population. Sci. Rep. 2017, 7, 2317. [Google Scholar] [CrossRef]
- Lechner, S.; Ferretti, L.; Schöning, C.; Kinuthia, W.; Willemsen, D.; Hasselmann, M. Nucleotide variability at its limit? Insights into the number and evolutionary dynamics of the sex-determining specificities of the honey bee Apis mellifera. Mol. Biol. Evolut. 2014, 31, 272–287. [Google Scholar] [CrossRef]
- Hasselmann, M.; Vekemans, X.; Pflugfelder, J.; Koeniger, N.; Koeniger, G.; Tingek, S.; Beye, M. Evidence for convergent nucleotide evolution and high allelic turnover rates at the complementary sex determiner gene of Western and Asian honeybees. Mol. Biol. Evolut. 2008, 25, 696–708. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Wu, X.; Yan, W.; Zeng, Z. Polymorphism analysis of csd gene in six Apis mellifera subspecies. Mol. Biol. Rep. 2012, 39, 3067–3071. [Google Scholar] [CrossRef]
- Laube, I.; Hird, H.; Brodmann, P.; Ullmann, S.; Schöne-Michling, M.; Chisholm, J.; Broll, H. Development of primer and probe sets for the detection of plant species in honey. Food Chem. 2010, 118, 979–986. [Google Scholar] [CrossRef]
- Ain, S.A.; Jesus, F.T.D.; Marchioro, G.M.; Araújo, E.D.D. Extraction of DNA from honey and its amplification by PCR for botanical identification. Food Sci. Technol. 2013, 33, 753–756. [Google Scholar]
- Bovo, S.; Utzeri, V.J.; Ribani, A.; Cabbri, R.; Fontanesi, L. Shotgun sequencing of honey DNA can describe honey bee derived environmental signatures and the honey bee hologenome complexity. Sci. Rep. 2020, 10, 1–17. [Google Scholar]
- Kek, S.P.; Chin, N.L.; Tan, S.W.; Yusof, Y.A.; Chua, L.S. Molecular identification of honey entomological origin based on bee mitochondrial 16S rRNA and COI gene sequences. Food Control 2017, 78, 150–159. [Google Scholar] [CrossRef]
- Utzeri, V.J.; Ribani, A.; Fontanesi, L. Authentication of honey based on a DNA method to differentiate Apis mellifera subspecies: Application to Sicilian honey bee (A. m. siciliana) and Iberian honey bee (A. m. iberiensis) honeys. Food Control 2018, 91, 294–301. [Google Scholar] [CrossRef]
- Prosser, S.W.; Hebert, P.D. Rapid identification of the botanical and entomological sources of honey using DNA metabarcoding. Food Chem. 2017, 214, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Soares, S.; Grazina, L.; Mafra, I.; Costa, J.; Pinto, M.A.; Duc, H.P.; Oliveira, M.B.P.; Amaral, J.S. Novel diagnostic tools for Asian (Apis cerana) and European (Apis mellifera) honey authentication. Food Res. Int. 2018, 105, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Châline, N.; Ratnieks, F.L.; Raine, N.E.; Badcock, N.S.; Burke, T. Non-lethal sampling of honey bee, Apis mellifera, DNA using wing tips. Apidologie 2004, 35, 311–318. [Google Scholar] [CrossRef]
- Mozes-Koch, R.; Slabezki, Y.; Efrat, H.; Kalev, H.; Kamer, Y.; Yakobson, B.A.; Dag, A. First detection in Israel of fluvalinate resistance in the varroa mite using bioassay and biochemical methods. Exp. Appl. Acarol. 2000, 24, 35–43. [Google Scholar] [CrossRef]
- Imkerbund, D. (Ed.) Methodenhandbuch der Arbeitsgmeinschaft Toleranzzucht (AGT); Deutschen Imkerbund: Kirchhain, Germany; Erlenstrasse: Basel, Switzerland, 2013. [Google Scholar]
- Kolics, É.; Mátyás, K.; Taller, J.; Specziár, A.; Kolics, B. Contact Effect Contribution to the High Efficiency of Lithium Chloride Against the Mite Parasite of the Honey Bee. Insects 2020, 11, 333. [Google Scholar] [CrossRef]
- Baudry, E.; Solignac, M.; Garnery, L.; Gries, M.; Cornuet, J.; Koeniger, N. Relatedness among honeybees (Apis mellifera) of a drone congregation. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1998, 265, 2009–2014. [Google Scholar] [CrossRef]
- Hasselmann, M.; Beye, M. Signatures of selection among sex-determining alleles of the honey bee. Proc. Natl. Acad. Sci. USA 2004, 101, 4888–4893. [Google Scholar] [CrossRef] [PubMed]
- Hasselmann, M.; Beye, M. Pronounced differences of recombination activity at the sex determination locus of the honeybee, a locus under strong balancing selection. Genetics 2006, 174, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D. Sex determination: Balancing selection in the honey bee. Curr. Biol. 2004, 14, R568–R569. [Google Scholar] [CrossRef] [PubMed]
- Beye, M.; Hunt, G.J.; Page, R.E.; Fondrk, M.K.; Grohmann, L.; Moritz, R.F.A. Unusually high recombination rate detected in the sex locus region of the honey bee (Apis mellifera). Genetics 1999, 153, 1701–1708. [Google Scholar]
- Liu, H.; Zhang, X.; Huang, J.; Chen, J.Q.; Tian, D.; Hurst, L.D.; Yang, S. Causes and consequences of crossing-over evidenced via a high-resolution recombinational landscape of the honey bee. Genome Biol. 2015, 16, 1–20. [Google Scholar] [CrossRef]


{kind=link}
| Country | Origin | Sample Type | Sample Code * |
|---|---|---|---|
| China | wild collected | bee, worker | Cw1 |
| China | wild collected | bee, worker | Cw2 |
| China | wild collected | bee, worker | Cw3.1 a |
| China | wild collected | bee, worker | Cw3.2 a |
| Hungary | private apiary, hive no 445/23 | bee, worker | Hw1.1 b |
| Hungary | private apiary, hive no 445/23 | bee, worker | Hw1.2 b |
| Hungary | private apiary, hive no 445/23 | bee, drone | Hd1.1 c |
| Hungary | private apiary, hive no 445/23 | bee, drone | Hd1.2 c |
| Hungary | private apiary, hive no 445/23 | bee, drone | Hd1.3 c |
| Hungary | private apiary (Kolics apiary) | bee, drone | Hd2 |
| Hungary | private apiary (Kolics apiary) | bee, drone | Hd3.1 d |
| Hungary | private apiary (Kolics apiary) | bee, drone | Hd3.2 d |
| Hungary | private apiary (Kolics apiary) | bee, drone | Hd4 |
| Hungary | private apiary (Kolics apiary) | bee, queen | Hq1.1 e |
| Hungary | private apiary (Kolics apiary) | bee, queen | Hq1.2 e |
| Hungary | private apiary (Kolics apiary) | bee, queen | Hq2.1 f |
| Hungary | private apiary (Kolics apiary) | bee, queen | Hq2.2 f |
| Hungary | private apiary (Kolics apiary) | bee, queen | Hq3.1 g |
| Hungary | private apiary (Kolics apiary) | bee, queen | Hq3.2 g |
| Hungary | private apiary (Kolics apiary) | bee, queen | Hq4 |
| China | local grocery | honey | Lao |
| China | local grocery | honey | Chi |
| Georgia | private apiary | honey | Gru |
| Japan | private apiary | honey | Jap |
| Hungary | private apiary | honey | Gel |
| Hungary | private apiary | honey | Szg |
| Hungary | private apiary | honey | Ves |
| Hungary | private apiary, hive no 445/23 | honey | 445/23 |
| Sample Codes * | Amino Acid Sequence of csd Alleles | Coverage Used ** | Coveragein in Total *** | Abundance of the Relevant Alleles |
|---|---|---|---|---|
| Cw1 | IISSLSNKTIHNNNNYKYNYNNNNYNNNYNNNCKKLYYNIINI | 4627 | 7848 | 71.34% |
| IISSLSNKTIHNNNNYKYNYNNNYNNNNNYNNYNNTNYKKLYYNINYI | 972 | |||
| Cw2 | IISSLSNNYNYNNNNYNNYNNNYNKKLYYNINYI | 4286 | 9280 | 80.23% |
| IISSLSNNYNYSNYNNYNNNNYNNYKKLYYNINYI | 3159 | |||
| Cw3.1 a | IISSLSNNYNYSNYNNYNNYNNNYNNYKKLYYNINYI | 11,699 | 23,013 | 78.38% |
| IISSLSNKTIHNNNNYKYNYNNNNNNYKNYNNYKKLYYNINYI | 6339 | |||
| Cw3.2 a | IISSLSNKTIHNNNNYKYNYNNNNNNYKNYNNYKKLYYNINYI | 7080 | 13,908 | 76.15% |
| IISSLSNNYNYSNYNNYNNYNNNYNNYKKLYYNINYI | 3511 | |||
| Hw1.1 b | IISSLSNKTIHNNNNYKYNYNNNCKKLYYNINYI | 15,779 | 31,092 | 81.03% |
| IISSLSNNYKYSNYNNYNNNNYNNNYNHYNNNYSKKLYYNINYI | 9416 | |||
| Hw1.2 b | IISSLSNKTIHNNNNYKYNYNNNCKKLYYNINYI | 13,505 | 30,441 | 81.30% |
| IISSLSNNYKYSNYNNYNNNNYNNNYNHYNNNYSKKLYYNINYI | 11,245 | |||
| Hd1.1 c | IISSLSNNYKYSNYNNYNNNNYNNNYNHYNNNYSKKLYYNINYI | 914 | 1325 | 68.98% |
| - | ||||
| Hd1.2 c | IISSLSNNYKYSNYNNYNNNNYNNNYNHYNNNYSKKLYYNINYI | 17,489 | 19,280 | 90.71% |
| - | ||||
| Hd1.3 c | IISSLSNNYKYSNYNNYNNNNYNNNYNHYNNNYSKKLYYNINYI | 139,098 | 149,456 | 93.1% |
| - | ||||
| Hd2 | IISSLSNNTIHNNNYKYNYNNNYNNYKKLYYNINYI | 4074 | 4300 | 94.74% |
| - | ||||
| Hd3.1 d | IISSLSNNTIHNNNYKYNYNNNYNNYKKLYYNINYI | 5431 | 5767 | 94.17% |
| - | ||||
| Hd3.2 d | IISSLSNNTIHNNNYKYNYNNNYNNYKKLYYNINYI | 11,949 | 12,708 | 94.03% |
| - | ||||
| Hd4 | IISSLSNNTIHNNNYKYNYNNNYNNYKKLYYNINYI | 14,886 | 15,985 | 93.12% |
| - | ||||
| Hq1.1 e | IISSLSNKTIHNNNNYNNNNNNYNNYNNYKKLYYNVINI | 8701 | 19,005 | 86.35% |
| IISSLSNNYKYSNYNNYNNNYNNYNNNYNNNYKKLYYNINYI | 7710 | |||
| Hq1.2 e | IISSLSNKTIHNNNNYNNNNNNYNNYNNYKKLYYNVINI | 8056 | 19,625 | 80.63% |
| IISSLSNNYKYSNYNNYNNNYNNYNNNYNNNYKKLYYNINYI | 7767 | |||
| Hq2.1 f | IISSLSNKTIHNNNNYNNNNYNNYKKLYYNIINI | 14,248 | 20,762 | 86.76% |
| IISSLSNNYNSNNYNNYNKYNYNNSKKLYYNINYI | 3765 | |||
| Hq2.2 f | IISSLSNKTIHNNNNYNNNNYNNYKKLYYNIINI | 10,176 | 13,493 | 91.28% |
| IISSLSNNYNSNNYNNYNKYNYNNSKKLYYNINYI | 2140 | |||
| Hq3.1 g | IISSLSNKTIHNNNNYNNNNNNYNNYNNYKKLYYNVINI | 7130 | 14,775 | 88.90% |
| IISSLSNNYKYSNYNNYNNNYNNYNNNYNNNYKKLYYNINYI | 6005 | |||
| Hq3.2 g | IISSLSNNYKYSNYNNYNNNYNNYNNNYNNNYKKLYYNINYI | 6437 | 14,346 | 89.50% |
| IISSLSNKTIHNNNNYNNNNNNYNNYNNYKKLYYNVINI | 6402 | |||
| Hq4 | IISSLSNKTIHNNNNYKPYYNINYI | 11,434 | 22,417 | 91.19% |
| IISSLSNNRNSNNYNNYNYKKLYYNINYI | 9007 |
| Sample | Subspecies | Amino Acid Sequence of the Hypervariable Region | Total Coverage * | Abundance in the Sample | NCBI Accession Number |
|---|---|---|---|---|---|
| Cw1_SA2, Jap_SA12 | ligustica | IISSLSNKTIHNNNNYKYNYNNNYNNNNNYNNYNNTNYKKLYYNINYI | 4577 (972 + 3605) | 12.4%, 1.5% | MK241931.1 identity to the most homologous existing sequence: 88% |
| Ves_SA4 | carnica | IISSLSNKTIHDNNNYKYNYNNNNNNYKNYNNYKKLYYNINYI | 1448 | 0.6% | MK241934.1 identity to the most homologous existing sequence: 98% |
| Ves_SA5 | carnica | IISSLSNNYNYSNYNNYNNYNKNYNNYKKLYYNINYI | 1317 | 0.5% | MK241935.1 identity to the most homologous existing sequence: 97% |
| Ves_SA6, Gru_SA4 | carnica/caucasica | IISSLSNKTIHNNNNYKYNYNNNNNYKNYNNYKKLYYNINYI | 2090 (1035 + 1055) | 0.4%, 0,4% | MK241936.1 identity to the most homologous existing sequence: 98% |
| Ves_SA7, Chi_SA3, Gru_SA3 | carnica/caucasica | IISSLSNKTIHNNNNYKYNYNNNNNYYKNYNNYKKLYYNINYI | 3482 (1027 + 1383 + 1072) | 0.4%, 0.7%, 0.4% | MK241937.1 identity to the most homologous existing sequence: 98% |
| SzG_SA4 | carnica | IISSLSNKTIHNNNNYKYNYNNNNYNNNNYKKLQYYNINYI | 22,572 | 3.5% | MK241933.1 identity to the most homologous existing sequence: 93% |
| Jap_SA14 | ligustica | IISSLSNKTIHNNNNYNNNNYNNYNNNYNNNNYNNYKKLYYNINYI | 1345 | 0.5% | MK241932.1 identity to the most homologous existing sequence: 96% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolics, É.; Parrag, T.; Házi, F.; Szepesi, K.; Heltai, B.; Mátyás, K.; Kutasy, B.; Virág, E.; Taller, J.; Orbán, L.; et al. An Alternative, High Throughput Method to Identify Csd Alleles of the Honey Bee. Insects 2020, 11, 483. https://doi.org/10.3390/insects11080483
Kolics É, Parrag T, Házi F, Szepesi K, Heltai B, Mátyás K, Kutasy B, Virág E, Taller J, Orbán L, et al. An Alternative, High Throughput Method to Identify Csd Alleles of the Honey Bee. Insects. 2020; 11(8):483. https://doi.org/10.3390/insects11080483
Chicago/Turabian StyleKolics, Éva, Tamás Parrag, Ferenc Házi, Kinga Szepesi, Botond Heltai, Kinga Mátyás, Barbara Kutasy, Eszter Virág, János Taller, László Orbán, and et al. 2020. "An Alternative, High Throughput Method to Identify Csd Alleles of the Honey Bee" Insects 11, no. 8: 483. https://doi.org/10.3390/insects11080483
APA StyleKolics, É., Parrag, T., Házi, F., Szepesi, K., Heltai, B., Mátyás, K., Kutasy, B., Virág, E., Taller, J., Orbán, L., & Kolics, B. (2020). An Alternative, High Throughput Method to Identify Csd Alleles of the Honey Bee. Insects, 11(8), 483. https://doi.org/10.3390/insects11080483