Temporal Variation in Genetic Composition of Migratory Helicoverpa Zea in Peripheral Populations



Abstract
1. Introduction
2. Materials and Methods
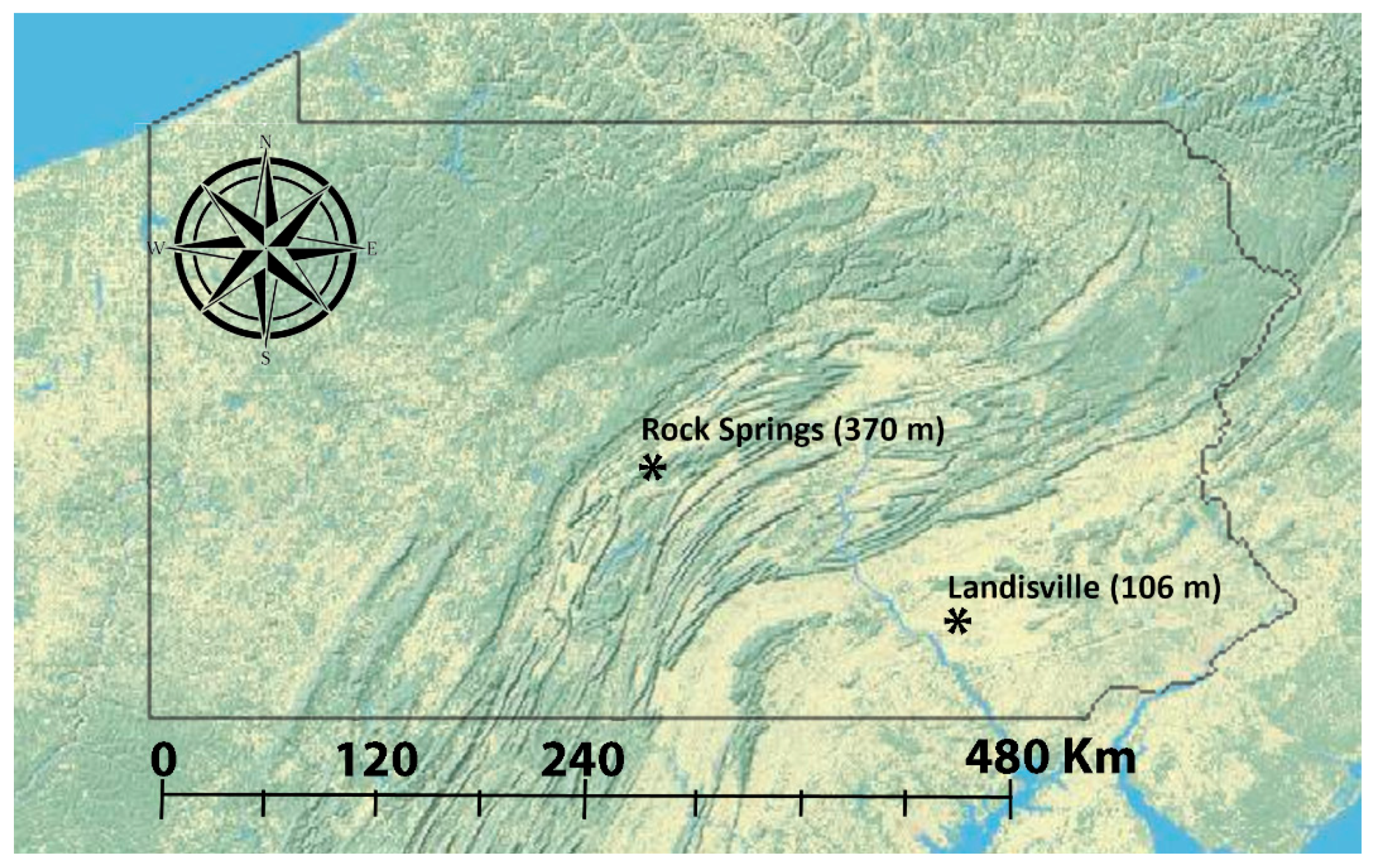
2.1. Sampling
2.2. Single Nucleotide Polymorphism Marker Development
2.3. Single Nucleotide Polymorphism (SNP) Analysis
2.4. Genetic Structure
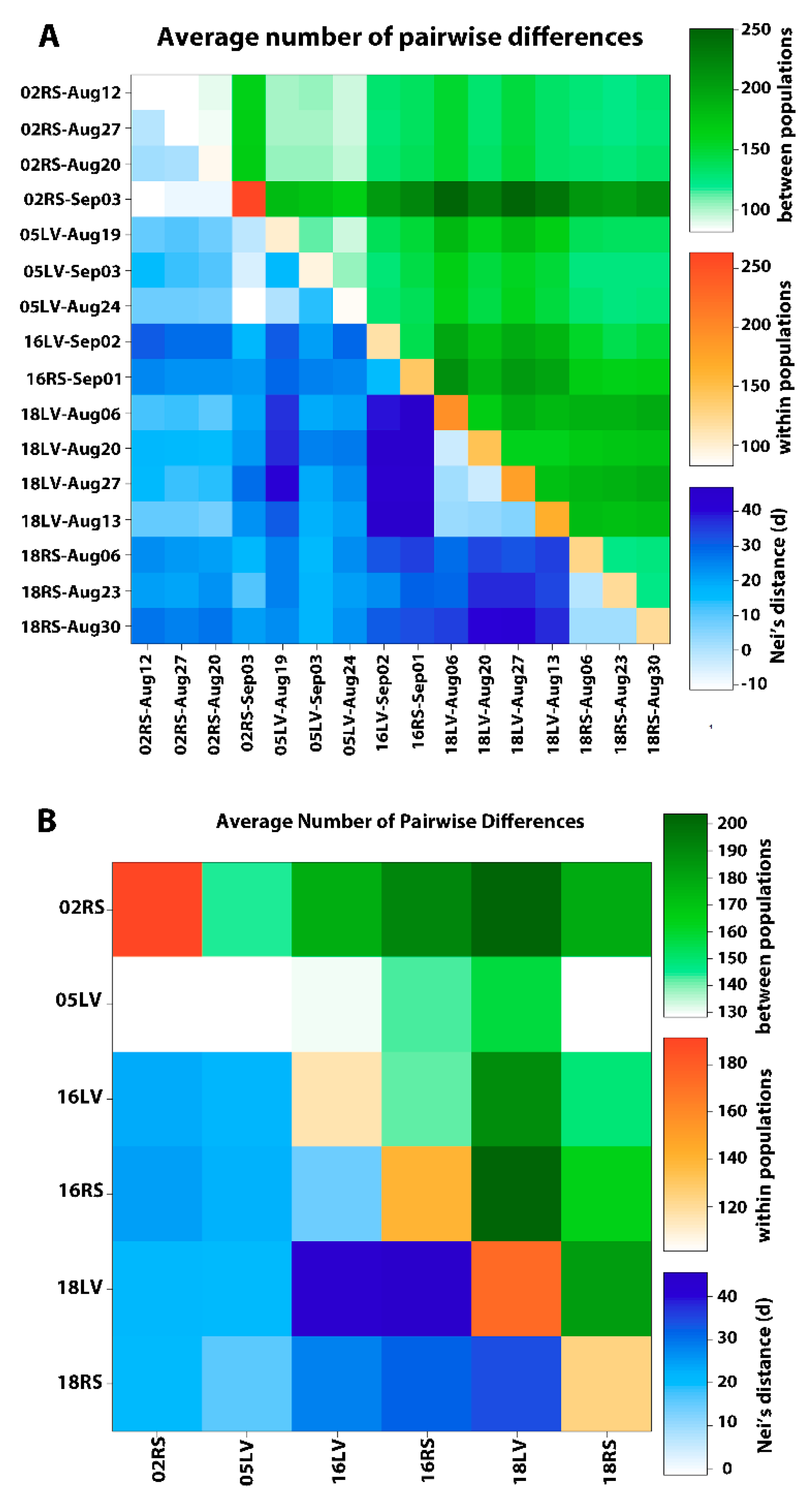
3. Results
3.1. SNP Discovery
3.2. SNP Analysis
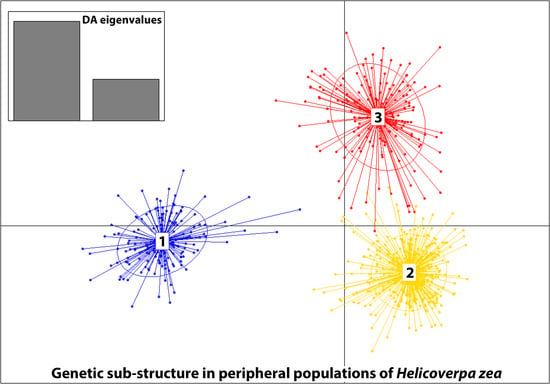
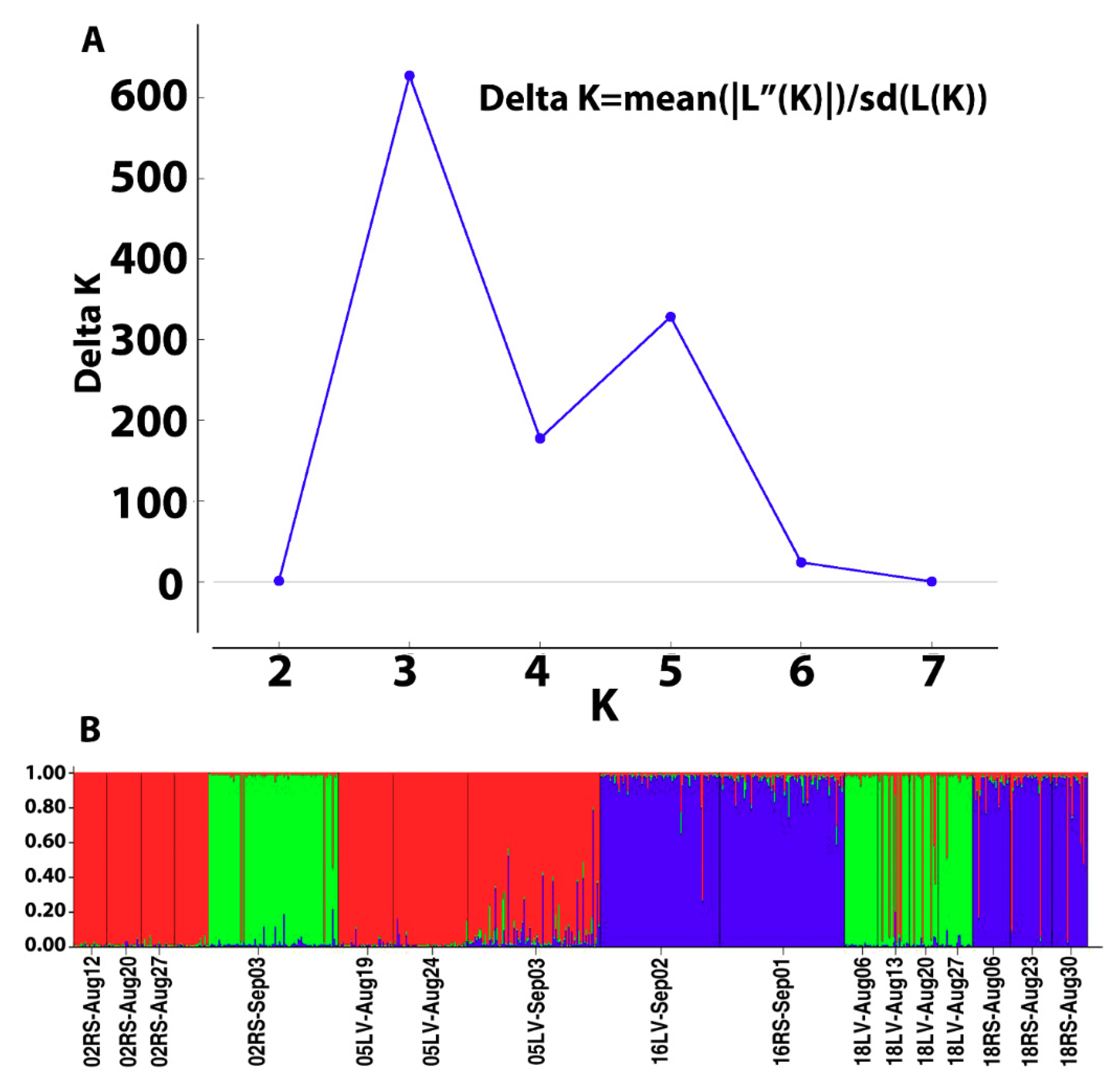
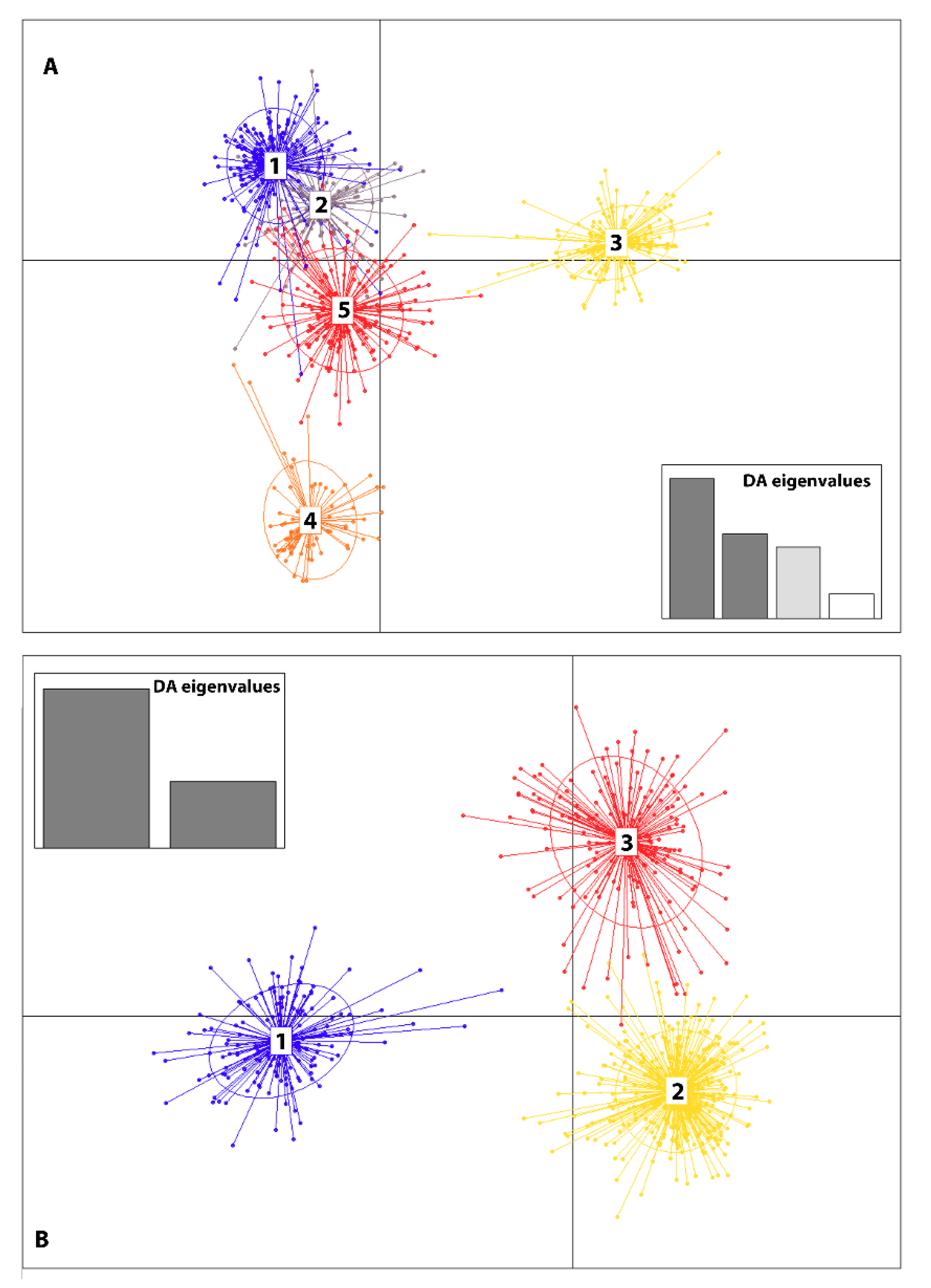
3.3. Genetic Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dingle, H.; Drake, V.A. What is migration? Bioscience 2007, 57, 113–121. [Google Scholar] [CrossRef]
- Slatkin, M. Gene flow and the geographic structure of natural populations. Science 1987, 236, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Eckert, C.; Samis, K.; Lougheed, S. Genetic variation across species’ geographical ranges: The central–marginal hypothesis and beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef]
- Hendry, A.P.; Day, T. Population structure attributable to reproductive time: Isolation by time and adaptation by time. Mol. Ecol. 2005, 14, 901–916. [Google Scholar] [CrossRef] [PubMed]
- Safriel, U.N.; Volis, S.; Kark, S. Core and peripheral populations and global climate change. Isr. J. Plant Sci. 1994, 42, 331–345. [Google Scholar] [CrossRef]
- Lenormand, T. Gene flow and the limits to natural selection. Trends Ecol. Evol. 2002, 17, 183–189. [Google Scholar] [CrossRef]
- Garant, D.; Forde, S.E.; Hendry, A.P. The multifarious effects of dispersal and gene flow on contemporary adaptation. Funct. Ecol. 2007, 21, 434–443. [Google Scholar] [CrossRef]
- García-Ramos, G.; Kirkpatrick, M. Genetic models of adaptation and gene flow in peripheral populations. Evolution 1997, 51, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.L.; Urban, M.C.; Bolnick, D.I.; Skelly, D.K. Microgeographic adaptation and the spatial scale of evolution. Trends Ecol. Evol. 2014, 29, 165–176. [Google Scholar] [CrossRef]
- Lingren, P.D.; Bryant, V.M., Jr.; Raulston, J.R.; Pendleton, M.; Westbrook, J.; Jones, G.D. Adult feeding host range and migratory activities of com earworm, cabbage looper, and celery looper (Lepidoptera: Noctuidae) moths as evidenced by attached pollen. J. Econ. Entomol. 1993, 86, 1429–1439. [Google Scholar] [CrossRef]
- Sudbrink, D.L., Jr.; Grant, J.F. Wild host plants of Helicoverpa zea and Heliothis virescens (Lepidoptera: Noctuidae) in eastern Tennessee. Environ. Entomol. 1995, 24, 1080–1085. [Google Scholar] [CrossRef]
- Kennedy, G.G.; Storer, N.P. Life systems of polyphagous arthropod pests in temporally unstable cropping systems. Annu. Rev. Entomol. 2000, 45, 467–493. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.R.; Threet, M. Cotton Insect Losses–2019. Available online: https://www.entomology.msstate.edu/resources/2019loss.php (accessed on 31 March 2020).
- Reisig, D.D.; Suits, R.; Burrack, H.; Bacheler, J.; Dunphy, J.E. Does florivory by Helicoverpa zea (Lepidoptera: Noctuidae) cause yield loss in soybeans? J. Econ. Entomol. 2017, 110, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.; Cook, D.; Catchot, A.; Gore, J.; Musser, F.; Stewart, S.; Kerns, D.; Lorenz, G.; Irby, J.; Golden, B. Evaluation of corn earworm, Helicoverpa zea (Lepidoptera: Noctuidae), economic injury levels in mid-south reproductive stage soybean. J. Econ. Entomol. 2016, 109, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Musser, F.R.; Catchot, A.L.; Davis, J.A.; Herbert, D.A.; Lorenz, G.M.; Reed, T.; Reisig, D.D.; Stewart, S.D. 2012 soybean insect losses in the southern US. Midsouth Entomol. 2013, 6, 12–24. [Google Scholar]
- Reay-Jones, F.P.F. Pest Status and Management of Corn Earworm (Lepidoptera: Noctuidae) in Field Corn in the United States. J. Integr. Pest Manag. 2019, 10, 19. [Google Scholar] [CrossRef]
- Olivi, B.; Gore, J.; Musser, F.; Catchot, A.; Cook, D. Impact of Simulated Corn Earworm (Lepidoptera: Noctuidae) Kernel Feeding on Field Corn Yield; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Reay-Jones, F.P.F.; Bessin, R.T.; Brewer, M.J.; Buntin, D.G.; Catchot, A.L.; Cook, D.R.; Flanders, K.L.; Kerns, D.L.; Porter, R.P.; Reisig, D.D. Impact of Lepidoptera (Crambidae, Noctuidae, and Pyralidae) pests on corn containing pyramided Bt traits and a blended refuge in the Southern United States. J. Econ. Entomol. 2016, 109, 1859–1871. [Google Scholar] [CrossRef]
- Bibb, J.L.; Cook, D.; Catchot, A.; Musser, F.; Stewart, S.D.; Leonard, B.R.; Buntin, G.D.; Kerns, D.; Allen, T.W.; Gore, J. Impact of corn earworm (Lepidoptera: Noctuidae) on field corn (Poales: Poaceae) yield and grain quality. J. Econ. Entomol. 2018, 111, 1249–1255. [Google Scholar] [CrossRef]
- Scholz, B.C.G.; Monsour, C.J.; Zalucki, M.P. An evaluation of selective Helicoverpa armigera control options in sweet corn. Aust. J. Exp. Agric. 1998, 38, 601–607. [Google Scholar] [CrossRef]
- Hendrix III, W.; Mueller, T.; Phillips, J.; Davis, O. Pollen as an indicator of long-distance movement of Heliothis zea (Lepidoptera: Noctuidae). Environ. Entomol. 1987, 16, 1148–1151. [Google Scholar] [CrossRef]
- Lingren, P.D.; Westbrook, J.K.; Bryant, V.M.; Raulston, J.R.; Esquivel, J.F.; Jones, G.D. Origin of Corn-Earworm (Lepidoptera, Noctuidae) Migrants as Determined by Citrus Pollen Markers and Synoptic Weather Systems. Environ. Entomol. 1994, 23, 562–570. [Google Scholar] [CrossRef]
- Fitt, G.P. The Ecology of Heliothis Species in Relation to Agroecosystems. Annu. Rev. Entomol. 1989, 34, 17–53. [Google Scholar] [CrossRef]
- Stadelbacher, E.A.; Furr, R.E.; Laster, M.L. Bollworms Lepidoptera-Noctuidae and Tobacco Budworms-Lepidoptera-Noctuidae Mortality of Adults Exposed to Insecticides on Cotton. J. Econ. Entomol. 1972, 65, 1682–1683. [Google Scholar] [CrossRef]
- Hartstack, A.; Lopez, J.; Muller, R.; Sterling, W.; King, E. Evidence of long range migration of Heliothis zea (Boddie) into Texas and Arkansas. Southwest. Entomol. (USA) 1982, 7, 188–201. [Google Scholar]
- Head, G.; Jackson, R.E.; Adamczyk, J.; Bradley, J.R.; Van Duyn, J.; Gore, J.; Hardee, D.D.; Leonard, B.R.; Luttrell, R.; Ruberson, J. Spatial and temporal variability in host use by Helicoverpa zea as measured by analyses of stable carbon isotope ratios and gossypol residues. J. Appl. Ecol. 2010, 47, 583–592. [Google Scholar] [CrossRef]
- USDA-ERS. Adoption of Genetically Engineered Crops in the U.S. 2019. Available online: https://www.ers.usda.gov/data-products/adoption-of-genetically-engineered-crops-in-the-us/recent-trends-in-ge-adoption.aspx (accessed on 31 March 2020).
- Mallet, J.; Korman, A.; Heckel, D.G.; King, P. Biochemical Genetics of Heliothis and Helicoverpa (Lepidoptera, Noctuidae) and Evidence for a Founder Event in Helicoverpa zea. Ann. Entomol. Soc. Am. 1993, 86, 189–197. [Google Scholar] [CrossRef]
- Perera, O.P.; Blanco, C.A. Microsatellite variation in Helicoverpa zea (Boddie) populations in the southern United States. Southwest. Entomol. 2011, 36, 271–286. [Google Scholar] [CrossRef]
- Reisig, D.D.; Huseth, A.S.; Bacheler, J.S.; Aghaee, M.-A.; Braswell, L.; Burrack, H.J.; Flanders, K.; Greene, J.K.; Herbert, D.A.; Jacobson, A. Long-term empirical and observational evidence of practical Helicoverpa zea resistance to cotton with pyramided Bt toxins. J. Econ. Entomol. 2018, 111, 1824–1833. [Google Scholar] [CrossRef]
- Suits, R.; Reisig, D.; Burrack, H. Feeding preference and performance of Helicoverpa zea (Lepidoptera: Noctuidae) larvae on various soybean tissue types. Fla. Entomol. 2017, 100, 162–167. [Google Scholar] [CrossRef]
- Reisig, D.D.; Kurtz, R. Bt resistance implications for Helicoverpa zea (Lepidoptera: Noctuidae) insecticide resistance management in the United States. Environ. Entomol. 2018, 47, 1357–1364. [Google Scholar] [CrossRef]
- Dively, G.P.; Venugopal, P.D.; Finkenbinder, C. Field-evolved resistance in corn earworm to Cry proteins expressed by transgenic sweet corn. PLoS ONE 2016, 11, e0169115. [Google Scholar] [CrossRef]
- Welch, K.L.; Unnithan, G.C.; Degain, B.A.; Wei, J.; Zhang, J.; Li, X.; Tabashnik, B.E.; Carriere, Y. Cross-resistance to toxins used in pyramided Bt crops and resistance to Bt sprays in Helicoverpa zea. J. Invertebr. Pathol. 2015, 132, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Tabashnik, B.E. ABCs of insect resistance to Bt. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Dively, G.P.; Kuhar, T.P.; Taylor, S.; Doughty, H.B.; Holmstrom, K.; Gilrein, D.; Nault, B.A.; Ingerson-Mahar, J.; Whalen, J.; Reisig, D.; et al. Sweet Corn Sentinel Monitoring for Lepidopteran Resistance to Bt Toxins. J. Econ. Entomol. 2020. in review. [Google Scholar]
- Perera, O.P.; Blanco, C.A.; Scheffler, B.E.; Abel, C.A. Characteristics of 13 polymorphic microsatellite markers in the corn earworm, Helicoverpa zea (Lepidoptera : Noctuidae). Mol. Ecol. Notes 2007, 7, 1132–1134. [Google Scholar] [CrossRef]
- Seymour, M.; Perera, O.P.; Fescemyer, H.W.; Jackson, R.E.; Fleischer, S.J.; Abel, C.A. Peripheral genetic structure of Helicoverpa zea indicates asymmetrical panmixia. Ecol. Evol. 2016, 6, 3198–3207. [Google Scholar] [CrossRef]
- Remington, C.L. The population genetics of insect introduction. Annu. Rev. Entomol. 1968, 13, 415–426. [Google Scholar] [CrossRef]
- Holt, R.D. Adaptive evolution in source-sink environments: Direct and indirect effects of density-dependence on niche evolution. Oikos 1996, 75, 182–192. [Google Scholar] [CrossRef]
- Hartstack, A.; Witz, J.; Buck, D. Moth traps for the tobacco budworm. J. Econ. Entomol. 1979, 72, 519–522. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Pearce, S.L.; Clarke, D.F.; East, P.D.; Elfekih, S.; Gordon, K.H.J.; Jermiin, L.S.; McGaughran, A.; Oakeshott, J.G.; Papanikolaou, A.; Perera, O.P.; et al. Genomic innovations, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive Helicoverpa pest species. BMC Biol. 2017, 15, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. An Exact Test for Population Differentiation. Evolution 1995, 49, 1280–1283. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J.; Raymond, M.; de Meeus, T.; Rousset, F. Testing differentiation in diploid populations. Genetics 1996, 144, 1933–1940. [Google Scholar] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Fisk, J.H. Karyotype and achiasmatic female meiosis in Helicoverpa armigera (Hübner) and H. punctigera (Wallengren)(Lepidoptera: Noctuidae). Genome 1989, 32, 967–971. [Google Scholar] [CrossRef]
- Kruglyak, L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 1999, 22, 139–144. [Google Scholar] [CrossRef]
- Carlson, C.S.; Eberle, M.A.; Rieder, M.J.; Yi, Q.; Kruglyak, L.; Nickerson, D.A. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 2004, 74, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.D.; Pritchard, J.K. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 2003, 4, 587–597. [Google Scholar] [CrossRef]
- Gerritsma, S.; Jalvingh, K.M.; van de Beld, C.; Beerda, J.; van de Zande, L.; Vrieling, K.; Wertheim, B. Natural and artificial selection for parasitoid resistance in Drosophila melanogaster leave different genetic signatures. Front. Genet. 2019, 10, 479. [Google Scholar] [CrossRef]
- Engström, P.G.; Sui, S.J.H.; Drivenes, Ø.; Becker, T.S.; Lenhard, B. Genomic regulatory blocks underlie extensive microsynteny conservation in insects. Genome Res. 2007, 17, 1898–1908. [Google Scholar] [CrossRef]
- Consortium, I.H. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851. [Google Scholar] [CrossRef]
- Wallberg, A.; Schoening, C.; Webster, M.T.; Hasselmann, M. Two extended haplotype blocks are associated with adaptation to high altitude habitats in East African honey bees. PLoS Genet. 2017, 13, e1006792. [Google Scholar] [CrossRef]
- Garud, N.R.; Petrov, D.A. Elevated linkage disequilibrium and signatures of soft sweeps are common in Drosophila melanogaster. Genetics 2016, 203, 863–880. [Google Scholar] [CrossRef] [PubMed]
- Garud, N.R.; Petrov, D.A. Elevation of linkage disequilibrium above neutral expectations in ancestral and derived populations of Drosophila melanogaster. Genetics 2016, 115, 184002. [Google Scholar]
- Rousset, F.; Raymond, M. Testing heterozygote excess and deficiency. Genetics 1995, 140, 1413–1419. [Google Scholar] [PubMed]
- Han, Q.; Caprio, M. Temporal and spatial patterns of allelic frequencies in cotton bollworm (Lepidoptera: Noctuidae). Environ. Entomol. 2002, 31, 462–468. [Google Scholar] [CrossRef]
- Sluss, T.P.; Sluss, E.S.; Graham, H.M.; Dubois, M. Allozyme Differences between Heliothis virescens (Lepidoptera-Noctuidae) and H. zea (Lepidoptera: Noctuidae). Ann. Entomol. Soc. Am. 1978, 71, 191–195. [Google Scholar] [CrossRef]
- Leite, N.A.; Alves-Pereira, A.; Corrêa, A.S.; Zucchi, M.I.; Omoto, C. Demographics and genetic variability of the new world bollworm (Helicoverpa zea) and the old world bollworm (Helicoverpa armigera) in Brazil. PLoS ONE 2014, 9, e113286. [Google Scholar] [CrossRef]
- Leite, N.A.; Correa, A.S.; Michel, A.P.; Alves-Pereira, A.; Pavinato, V.A.C.; Zucchi, M.I.; Omoto, C. Pan-American similarities in genetic structures of Helicoverpa armigera and Helicoverpa zea (Lepidoptera: Noctuidae) with implications for hybridization. Environ. Entomol. 2017, 46, 1024–1034. [Google Scholar] [CrossRef]
- Farrow, R.A.; Daly, J. Long-range movements as an adaptive strategy in the genus Heliothis (Lepidoptera, Noctuidae)-a review of its occurrence and detection in 4 pest species. Aust. J. Zool. 1987, 35, 1–24. [Google Scholar] [CrossRef]
- Westbrook, J.K.; López, J.D. Long-Distance Migration in Helicoverpa zea: What We Know and Need to Know. Southwest. Entomol. 2010, 35, 355–360. [Google Scholar] [CrossRef]
- Westbrook, J.K. Noctuid migration in Texas within the nocturnal aeroecological boundary layer. Integr. Comp. Biol. 2008, 48, 99–106. [Google Scholar] [CrossRef]
- Nibouche, S.; Buès, R.; Toubon, J.-F.; Poitout, S. Allozyme polymorphism in the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae): Comparison of African and European populations. Heredity 1998, 80, 438–445. [Google Scholar] [CrossRef]
- Asokan, R.; Nagesha, S.; Manamohan, M.; Krishnakumar, N.; Mahadevaswamy, H.; Rebijith, K.; Prakash, M.; Chandra, G.S. Molecular diversity of Helicoverpa armigera hubner (Noctuidae: Lepidoptera) in India. Orient. Insects 2012, 46, 130–143. [Google Scholar] [CrossRef]
- Endersby, N.M.; Hoffmann, A.A.; McKechnie, S.W.; Weeks, A.R. Is there genetic structure in populations of Helicoverpa armigera from Australia? Entomol. Exp. Appl. 2007, 122, 253–263. [Google Scholar] [CrossRef]
- Zhou, X.F.; Faktor, O.; Applebaum, S.W.; Coll, M. Population structure of the pestiferous moth Helicoverpa armigera in the Eastern Mediterranean using RAPD analysis. Heredity 2000, 85, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Vijaykumar, F.B.; Krishnareddy, K.B.; Kuruvinashetti, M.S.; Patil, B.V. Genetic differentiation among cotton bollworm, Helicoverpa armigera (Hübner) populations of south Indian cotton ecosystems using mitochondrial DNA markers. Ital. J. Zool. 2008, 75, 437–443. [Google Scholar] [CrossRef][Green Version]
- Deepa, M.; Srivastava, C. Genetic diversity in Helicoverpa armigera (Hübner) from different agroclimatic zones of India using RAPD markers. J. Food Legumes 2011, 24, 313–316. [Google Scholar]
- Scott, K.D.; Wilkinson, K.S.; Merritt, M.A.; Scott, L.J.; Lange, C.L.; Schutze, M.K.; Kent, J.K.; Merritt, D.J.; Grundy, P.R.; Graham, G.C. Genetic shifts in Helicoverpa armigera Hübner (Lepidoptera: Noctuidae) over a year in the Dawson/Callide Valleys. Aust. J. Agric. Res. 2003, 54, 739–744. [Google Scholar] [CrossRef]
- Scott, K.D.; Wilkinson, K.S.; Lawrence, N.; Lange, C.L.; Scott, L.J.; Merritt, M.A.; Lowe, A.J.; Graham, G.C. Gene-flow between populations of cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae) is highly variable between years. Bull. Entomol. Res. 2005, 95, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, J.; Fleischer, S.; Jairam, S.; Meagher, R.; Nagoshi, R. Multigenerational migration of fall armyworm, a pest insect. Ecosphere 2019, 10, e02919. [Google Scholar] [CrossRef]
- Westbrook, J.K.; Nagoshi, R.N.; Meagher, R.L.; Fleischer, S.J.; Jairam, S. Modeling seasonal migration of fall armyworm moths. Int. J. Biometeorol. 2016, 60, 255–267. [Google Scholar] [CrossRef]
- Showers, W.B. Migratory ecology of the black cutworm. Annu. Rev. Entomol. 1997, 42, 393–425. [Google Scholar] [CrossRef] [PubMed]
- Isard, S.A.; Gage, S.H. Flow of Life in the Atmosphere: An Airscape Approach to Understanding Invasive Organisms; Michigan State University Press: East Lansing, MI, USA, 2001; p. 240. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | GPS Coordinates | Name of Putative Population | Collection Year | Collection Date | Sample Size |
|---|---|---|---|---|---|
| Rock Springs, PA | 40°42′38.1″ N 77°57′52.2″ W | 02RS-Aug12 | 2002 | August 12 | 25 |
| 02RS-Aug20 | 2002 | August 20 | 24 | ||
| 02RS-Aug27 | 2002 | August 27 | 24 | ||
| 02RS-Sep03 | 2002 | September 03 | 117 | ||
| 16RS-Sep01 | 2016 | September 01 | 95 | ||
| 18RS-Aug06 | 2018 | August 06 | 33 | ||
| 18RS-Aug23 | 2018 | August 23 | 32 | ||
| 18RS-Aug30 | 2018 | August 30 | 30 | ||
| Landisville, PA | 40°07′04.7″ N 76°25′30.5″ W | 05LV-Aug19 | 2005 | August 19 | 39 |
| 05LV-Aug24 | 2005 | August 24 | 56 | ||
| 05LV-Sep03 | 2005 | September 3 | 95 | ||
| 16LV-Sep02 | 2016 | September 2 | 95 | ||
| 18LV-Aug06 | 2018 | August 6 | 24 | ||
| 18LV-Aug13 | 2018 | August 13 | 24 | ||
| 18LV-Aug20 | 2018 | August 20 | 23 | ||
| 18LV-Aug27 | 2018 | August 27 | 24 | ||
| Total | 760 |
| Population Number | Population Name | N | FIS | p |
|---|---|---|---|---|
| 1 | 02RS-Aug12 | 23 | 0.1972 | 0.084 |
| 2 | 02RS-Aug20 | 24 | 0.2330 | 0.0551 |
| 3 | 02RS-Aug27 | 23 | 0.1734 | 0.1235 |
| 4 | 02RS-Sep03 | 114 | −0.0350 | 0.6418 |
| 5 | 05LV-Aug19 | 35 | −0.3623 | 0.9814 |
| 6 | 05LV-Aug24 | 52 | * 0.1851 | 0.0270 |
| 7 | 05LV-Sep03 | 92 | * 0.1467 | 0.0285 |
| 8 | 16LV-Sep02 | 83 | 0.0019 | 0.5021 |
| 9 | 16RS-Sep01 | 87 | 0.1325 | 0.0763 |
| 10 | 18LV-Aug06 | 23 | 0.0312 | 0.4629 |
| 11 | 18LV-Aug13 | 22 | −0.0267 | 0.5750 |
| 12 | 18LV-Aug20 | 20 | −0.0640 | 0.6333 |
| 13 | 18LV-Aug27 | 24 | −0.0440 | 0.6034 |
| 14 | 18RS-Aug06 | 26 | 0.0247 | 0.4699 |
| 15 | 18RS-Aug23 | 29 | 0.1176 | 0.2498 |
| 16 | 18RS-Aug30 | 25 | 0.0988 | 0.2839 |
| Type of Grouping | Variance Component | % Total Variance | Φ-Statistic | p |
|---|---|---|---|---|
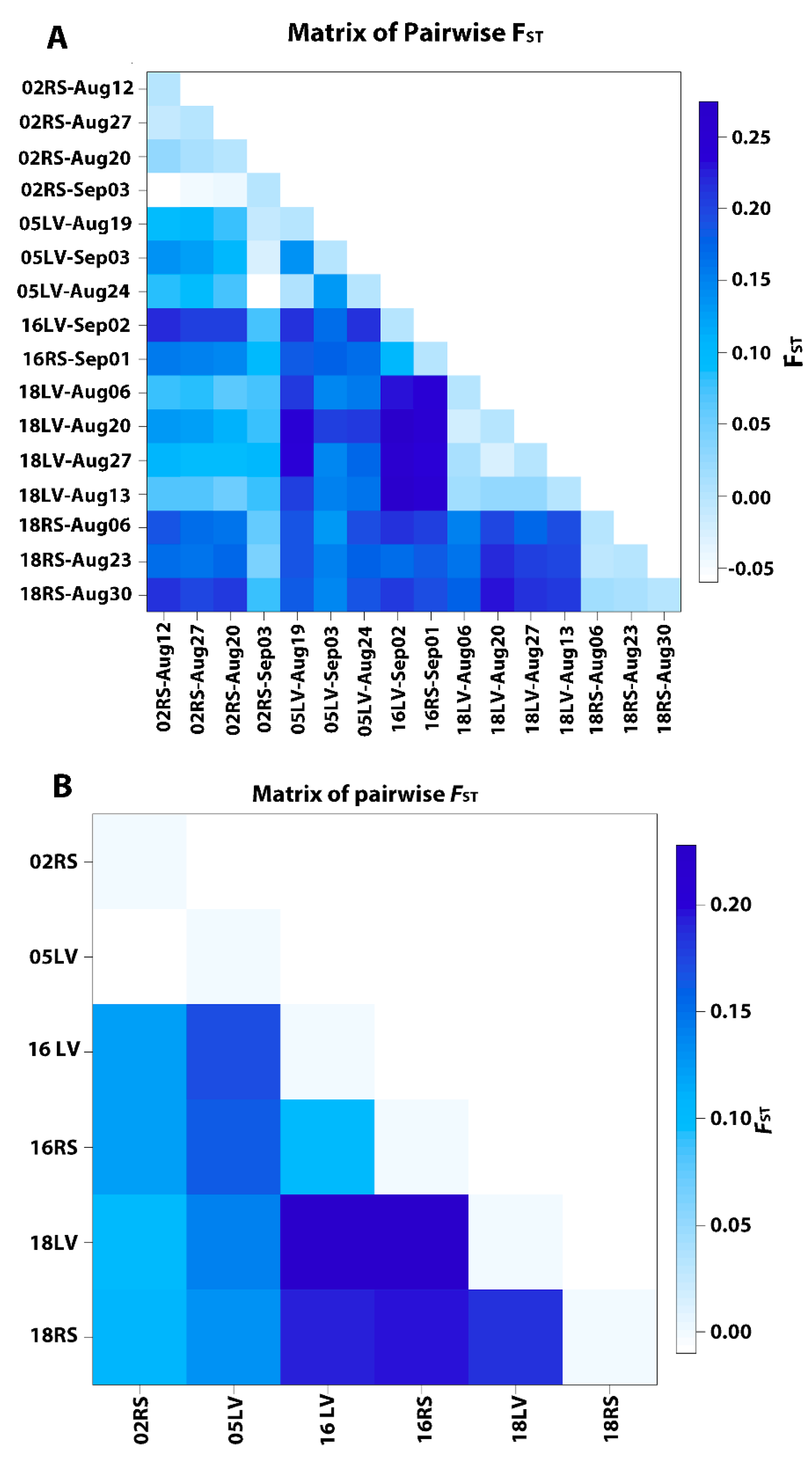
| 1. All populations Group 1 [All populations | Among populations | 4.89 | FST = 0.0489 | <0.000001 |
| 2. Grouped by collection Year Group 1 [02RS1, 02RS2] Group 2 [05LV1, 05LV2] Group 3 [16LV, 16RS3] Group 4 [18LV, 18RS] | Among groups Among populations within groups | 4.84 7.29 | FCT =−0.04836 FST = 0.078669 | =0.07871 <0.000001 |
| 3. Grouped by collection date Group 1 [02RS-Aug12, 18LV-Aug13 18RS-Aug06, 18LV-Aug06] Group 2 [02RS-Aug20, 18LV-Aug20 05LV-Aug19, 05LV-Aug24 18RS-Aug23] Group 3 [18RS-Aug30, 16RS-Sep01 02RS-Sep03, 16LV-Sep02, 05LV-Sep03] Group 4 [02RS-Aug27, 18LV-Aug27] | Among groups Among populations within groups | 1.85 10.34 | FCT = 0.01847 FST = 0.10539 | =0.11101 <0.000001 |
| 4. Grouped by the proportion of Cluster assignment of genotypes in the collection based on STRUCTURE results (Figure 3A,B) | ||||
| Group 1 [C1-02RS2, C1-18LV] Group 2 [C2-02R1, C2-02R2 05LV-Aug19, 05LV-Aug24 18RS-Aug23] Group 3 [C3-16LV, C3-16RS. C3-18RS] | Among groups Among populations within groups | 0.45 8.21 | IFCT = 0.00583 FST = 0.10799 | =0.41673 <0.000001 |
| 5. Grouped by genetic cluster assignment Group 1[Cluster1, Cluster 2, Cluster 3] | Among clusters | 4.85 | FST = 0.05283 | <0.000001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, O.P.; Fescemyer, H.W.; Fleischer, S.J.; Abel, C.A. Temporal Variation in Genetic Composition of Migratory Helicoverpa Zea in Peripheral Populations. Insects 2020, 11, 463. https://doi.org/10.3390/insects11080463
Perera OP, Fescemyer HW, Fleischer SJ, Abel CA. Temporal Variation in Genetic Composition of Migratory Helicoverpa Zea in Peripheral Populations. Insects. 2020; 11(8):463. https://doi.org/10.3390/insects11080463
Chicago/Turabian StylePerera, Omaththage P., Howard W. Fescemyer, Shelby J. Fleischer, and Craig A. Abel. 2020. "Temporal Variation in Genetic Composition of Migratory Helicoverpa Zea in Peripheral Populations" Insects 11, no. 8: 463. https://doi.org/10.3390/insects11080463
APA StylePerera, O. P., Fescemyer, H. W., Fleischer, S. J., & Abel, C. A. (2020). Temporal Variation in Genetic Composition of Migratory Helicoverpa Zea in Peripheral Populations. Insects, 11(8), 463. https://doi.org/10.3390/insects11080463