Non-Destructive Genotyping of Honeybee Queens to Support Selection and Breeding


Abstract
Simple Summary
Abstract
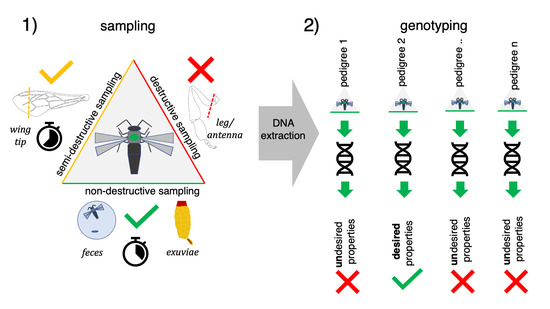
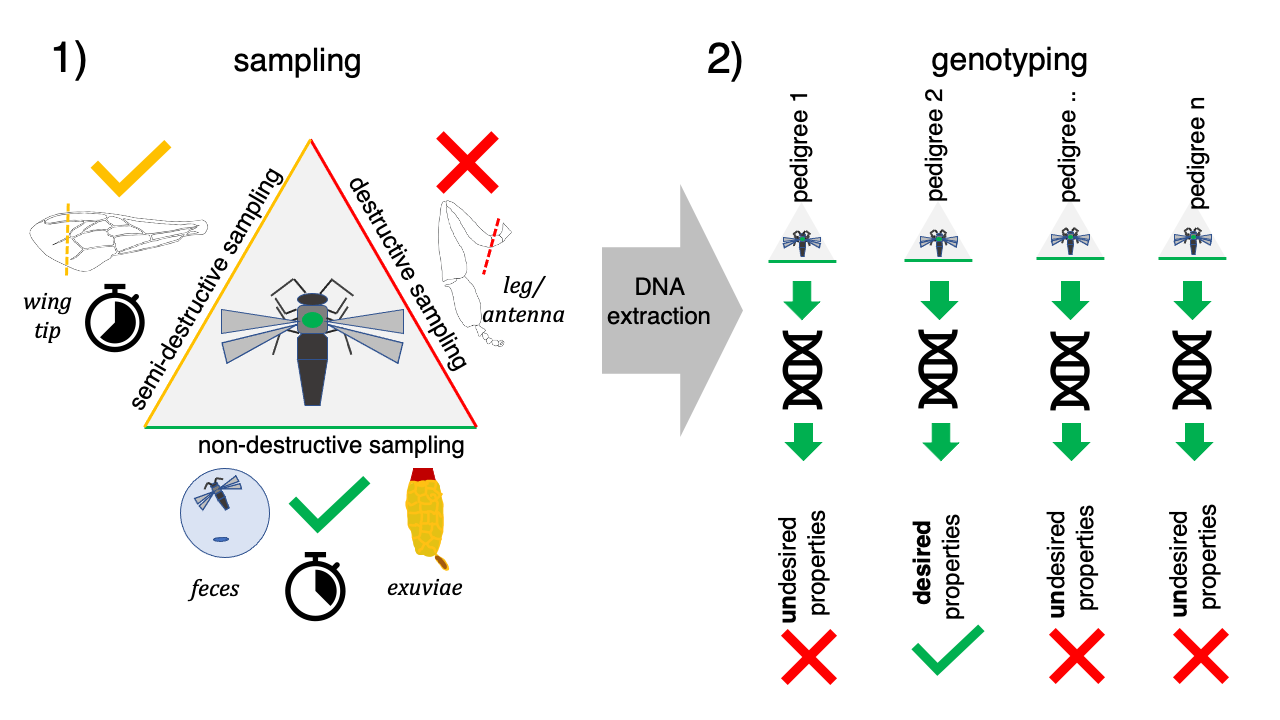
1. Introduction
2. Materials and Methods
2.1. Samples Information
2.2. Feces Sampling
2.3. Sampling of Exuviae
2.4. Tissue Sampling
2.5. DNA Extraction
2.6. PCR Amplification, Purification, and Sequencing of Specific Fragments
2.7. Phylogenetic Analysis
2.8. Genotyping Using dCAPS Protocol
3. Results
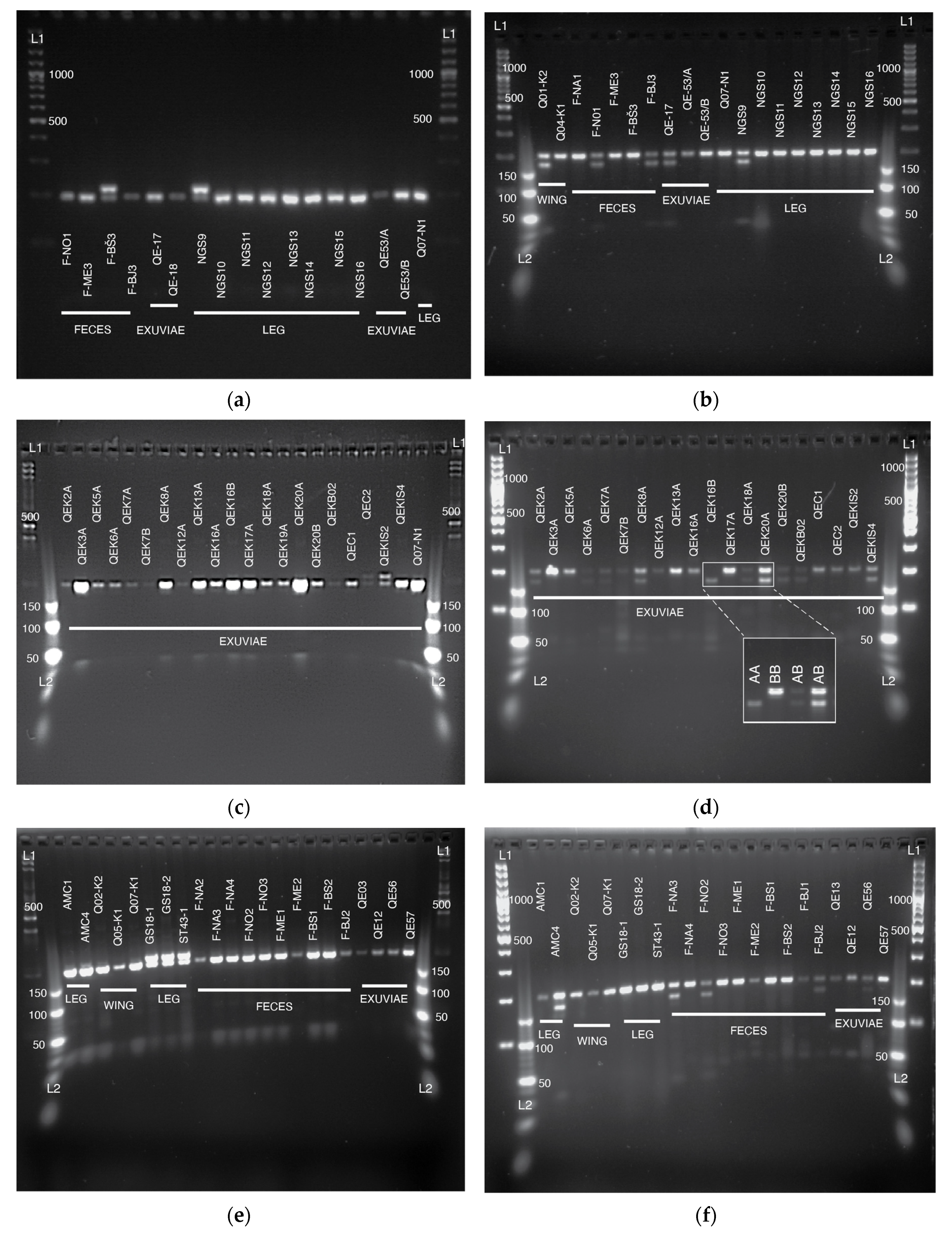
3.1. Sampling from Non-Destructive Sources and Extraction of DNA
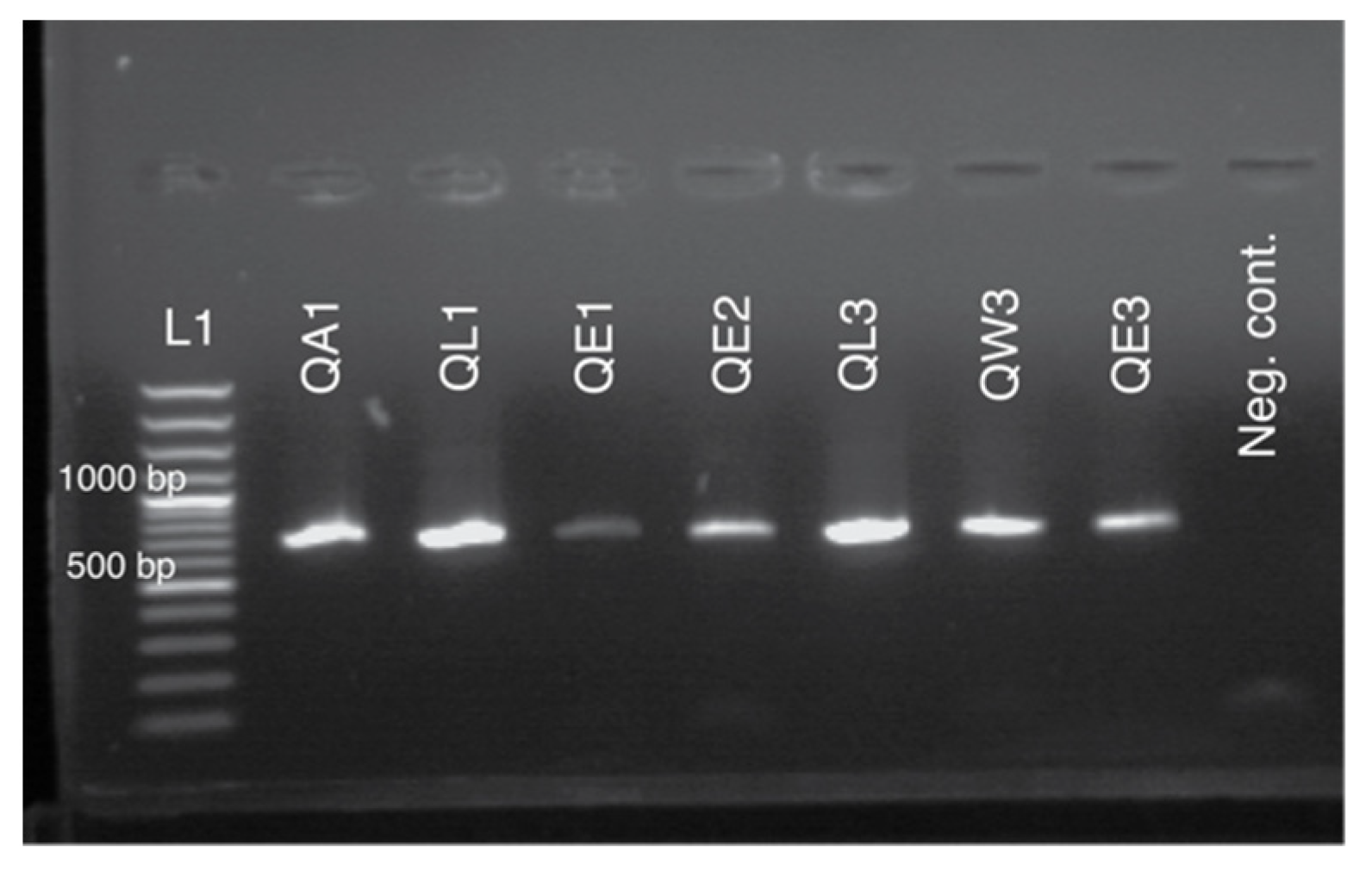
3.2. Sequencing and Genotyping Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holloway, B.; Tarver, M.R.; Rinderer, T.E. Fine mapping identifies significantly associating markers for resistance to the honey bee brood fungal disease, Chalkbrood. J. Apic. Res. 2013, 52, 134–140. [Google Scholar] [CrossRef]
- Spötter, A.; Gupta, P.; Mayer, M.; Reinsch, N.; Bienefeld, K. Genome-Wide Association Study of a Varroa-Specific Defense Behavior in Honeybees (Apis mellifera). J. Hered. 2016, 107, 220. [Google Scholar] [CrossRef] [PubMed]
- Bixby, M.; Baylis, K.; Hoover, S.E.; Currie, R.W.; Melathopoulos, A.P.; Pernal, S.F.; Foster, L.J.; Guarna, M.M. A Bio-Economic Case Study of Canadian Honey Bee (Hymenoptera: Apidae) Colonies: Marker-Assisted Selection (MAS) in Queen Breeding Affects Beekeeper Profits. J. Econ. Entomol. 2017, 110, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Wragg, D.; Marti-Marimon, M.; Basso, B.; Bidanel, J.-P.; Labarthe, E.; Bouchez, O.; Le Conte, Y.; Vignal, A. Whole-genome resequencing of honeybee drones to detect genomic selection in a population managed for royal jelly. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Meixner, M.D.; Pinto, M.A.; Bouga, M.; Kryger, P.; Ivanova, E.; Fuchs, S. Standard methods for characterising subspecies and ecotypes of Apis mellifera. J. Apic. Res. 2013, 52, 1–28. [Google Scholar] [CrossRef]
- Pinto, M.A.; Henriques, D.; Chávez-Galarza, J.; Kryger, P.; Garnery, L.; Van Der Zee, R.; Dahle, B.; Soland-Reckeweg, G.; De La Rúa, P.; Olio, R.D.; et al. Genetic integrity of the Dark European honey bee (Apis mellifera mellifera) from protected populations: A genome-wide assessment using SNPs and mtDNA sequence data. J. Apic. Res. 2014, 53, 269–278. [Google Scholar] [CrossRef]
- Behura, S.K. Molecular marker systems in insects: Current trends and future avenues. Mol. Ecol. 2006, 15, 3087–3113. [Google Scholar] [CrossRef]
- Evans, J.D.; Schwarz, R.S.; Chen, Y.; Budge, G.; Cornman, R.S.; De La Rua, P.; De Miranda, J.R.; Foret, S.; Foster, L.; Gauthier, L.; et al. Standard methods for molecular research in Apis mellifera. J. Apic. Res. 2013, 52, 1–54. [Google Scholar] [CrossRef]
- Butler, C.G. The process of queen supersedure in colonies of honeybees (Apis mellifera Linn.). Insectes Sociaux 1957, 4, 211–223. [Google Scholar] [CrossRef]
- Wedmore, E.B. A Manual of Bee-Keeping for English-Speaking Bee-Keepers; Bridgewater: Westport, CT, USA, 1984. [Google Scholar]
- Gerula, D.; Bienkowska, M. Performance of artificially inseminated honeybee queens that have bodily injuries. J. Apic. Sci. 2008, 52, 13–20. [Google Scholar]
- Jones, J.C.; Du, Z.G.; Bernstein, R.; Meyer, M.; Hoppe, A.; Schilling, E.; Ableitner, M.; Juling, K.; Dick, R.; Strauss, A.S.; et al. Tool for genomic selection and breeding to evolutionary adaptation: Development of a 100K single nucleotide polymorphism array for the honey bee. Ecol. Evol. 2020, 10, 6246–6256. [Google Scholar] [CrossRef] [PubMed]
- Châline, N.; Ratnieks, F.L.W.; Arnold, G.; Papin, C. Patriline differences in emergency queen rearing in the honey bee, Apis mellifera. Insectes Sociaux 2003, 50, 234–236. [Google Scholar] [CrossRef]
- Gregory, P.G.; Rinderer, T.E. Non-destructive sources of DNA used to genotype honey bee (Apis mellifera) queens. Entomol. Exp. Appl. 2004, 111, 173–177. [Google Scholar] [CrossRef]
- Gould, E.M.; Taylor, M.A.; Holmes, S.J. A more consistent method for extracting and amplifying DNA from bee wings. Apidologie 2011, 42, 721–727. [Google Scholar] [CrossRef]
- Facchini, E.; Rizzi, R.M.; Chessa, S. DNA extraction from wings as a suitable approach for queen bees genotyping. Int. J. Health Anim. Sci. Food Saf. 2018, 5. [Google Scholar] [CrossRef]
- Châline, N.; Ratnieks, F.L.W.; Raine, N.E.; Badcock, N.S.; Burke, T. Non-lethal sampling of honey bee, Apis mellifera, DNA using wing tips. Apidologie 2004, 35, 311–318. [Google Scholar] [CrossRef]
- Kolics, É.; Parrag, T.; Házi, F.; Szepesi, K.; Heltai, B.; Mátyás, K.; Kutasy, B.; Virág, E.; Taller, J.; Orbán, L.; et al. An Alternative, High Throughput Method to Identify Csd Alleles of the Honey Bee. Insects 2020, 11, 483. [Google Scholar] [CrossRef]
- Bertholf, L.M. The Moults of the Honeybee. J. Econ. Entomol. 1925, 18, 380–384. [Google Scholar] [CrossRef]
- Watts, P.C.; Thompson, D.J.; Kemp, S.J. Exuviae as a reliable source of DNA for population-genetic analysis of odonates. Odonatologica 2005, 34, 183–187. [Google Scholar]
- Nguyen, H.Q.; Kim, Y.I.; Borzée, A.; Jang, Y. Efficient isolation method for high-quality genomic DNA from cicada exuviae. Ecol. Evol. 2017, 7, 8161–8169. [Google Scholar] [CrossRef]
- Su, S.; Albert, Š.; Zhang, S.; Maier, S.; Chen, S.; Du, H.; Tautz, J. Non-destructive genotyping and genetic variation of fanning in a honey bee colony. J. Insect Physiol. 2007, 53, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Scriven, J.J.; Woodall, L.C.; Goulson, D. Nondestructive DNA sampling from bumblebee faeces. Mol. Ecol. Resour. 2013, 13, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Nboyine, J.A.; Boyer, S.; Saville, D.J.; Wratten, S.D. Identifying plant DNA in the faeces of a generalist insect pest to inform trap cropping strategy. Agron. Sustain. Dev. 2019, 39, 57. [Google Scholar] [CrossRef]
- Ide, T.; Kanzaki, N.; Ohmura, W.; Okabe, K. Molecular Identification of an Invasive Wood-Boring Insect Lyctus brunneus (Coleoptera: Bostrichidae: Lyctinae) Using Frass by Loop-Mediated Isothermal Amplification and Nested PCR Assays. J. Econ. Entomol. 2016, 109, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Kozmus, P.; Podgoršek, P.; Škerl, M.I.S.; Prešern, J.; Metelko, M.; Hrastelj, M. Rejski Program za Kranjsko Čebelo (Apis mellifera Carnica); Čebelarska zveza Slovenije: Lukovica pri Domžalah, Slovenia, 2018; Available online: http://www.czs.si/Upload/files/RP_2018%20(oddano%20na%20MKGP)_JP.doc (accessed on 1 September 2020).
- Elias-Neto, M.; Soares, M.P.M.; Bitondi, M.M.G. Changes in integument structure during the imaginal molt of the honey bee. Apidologie 2008, 40, 29–39. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Garnery, L.; Franck, P.; Baudry, E.; Vautrin, D.; Cornuet, J.-M.; Solignac, M. Genetic diversity of the west European honey bee (Apis mellifera mellifera and A. m. iberica) I. Mitochondrial DNA. Genet. Sel. Evol. 1998, 30 (Suppl. S1), S31. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Spötter, A.; Gupta, P.; Nürnberg, G.; Reinsch, N.; Bienefeld, K. Development of a 44K SNP assay focussing on the analysis of a varroa-specific defence behaviour in honey bees (Apis mellifera carnica): SNP assay for varroa tolerance in honey bees. Mol. Ecol. Resour. 2012, 12, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Tsuruda, J.M.; Harris, J.W.; Bourgeois, L.; Danka, R.G.; Hunt, G.J. High-Resolution Linkage Analyses to Identify Genes That Influence Varroa Sensitive Hygiene Behavior in Honey Bees. PLoS ONE 2012, 7, e48276. [Google Scholar] [CrossRef] [PubMed]
- Neff, M.M.; Turk, E.; Kalishman, M. Web-based primer design for single nucleotide polymorphism analysis. Trends Genet. 2002, 18, 613–615. [Google Scholar] [CrossRef]
- Alburaki, M.; Moulin, S.; Legout, H.; Alburaki, A.; Garnery, L. Mitochondrial structure of Eastern honeybee populations from Syria, Lebanon and Iraq. Apidologie 2011, 42, 628. [Google Scholar] [CrossRef]
- Walsh, P.S.; Metzger, D.A.; Higuchi, R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 1991, 10, 506–513. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. Hebert. bold: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Muñoz, I.; Henriques, D.; Johnston, J.S.; Chávez-Galarza, J.; Kryger, P.; Pinto, M.A. Reduced SNP Panels for Genetic Identification and Introgression Analysis in the Dark Honey Bee (Apis mellifera mellifera). PLoS ONE 2015, 10, e0124365. [Google Scholar] [CrossRef]
- Traynor, K.S.; Mondet, F.; de Miranda, J.R.; Techer, M.; Kowallik, V.; Oddie, M.A.Y.; Chantawannakul, P.; McAfee, A. Varroa destructor: A Complex Parasite, Crippling Honey Bees Worldwide. Trends Parasitol. 2020, 36, 592–606. [Google Scholar] [CrossRef]
- Guarna, M.M.; Hoover, S.E.; Huxter, E.; Higo, H.; Moon, K.-M.; Domanski, D.; Bixby, M.E.F.; Melathopoulos, A.P.; Ibrahim, A.; Peirson, M.; et al. Peptide biomarkers used for the selective breeding of a complex polygenic trait in honey bees. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Baumbusch, L.O.; Sundal, I.K.; Hughes, D.W.; Galau, G.A.; Jakobsen, K.S. Efficient protocols for CAPS-based mapping in Arabidopsis. Plant Mol. Biol. Rep. 2001, 19, 137–149. [Google Scholar] [CrossRef]
- Singh, R.; Deb, R.; Singh, U.; Raja, T.; Alex, R.; Kumar, S.; Chakraborti, S.; Sengar, G.; Sharma, S. Single tube tetraplex PCR based screening of a SNP at exon 14 region of bovine ITGB6 among different Zebu breeds. Meta Gene 2015, 3, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Syromyatnikov, M.; Borodachev, A.; Kokina, A.; Popov, V. A Molecular Method for the Identification of Honey Bee Subspecies Used by Beekeepers in Russia. Insects 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.; Zhou, Y.; Zhang, Q.; Niu, C.; Zhu, Z. Development of an easy and cost-effective method for non-invasive genotyping of insects. PLoS ONE 2019, 14, e0216998. [Google Scholar] [CrossRef] [PubMed]
- Monroe, E.M.; Lynch, C.; Soluk, D.A.; Britten, H.B. Nonlethal Tissue Sampling Techniques and Microsatellite Markers Used for First Report of Genetic Diversity in Two Populations of the Endangered Somatochlora hineana (Odonata: Corduliidae). Ann. Entomol. Soc. Am. 2010, 103, 1012–1017. [Google Scholar] [CrossRef]
- Frantzen, M.A.J.; Silk, J.B.; Ferguson, J.W.H.; Wayne, R.K.; Kohn, M.H. Empirical evaluation of preservation methods for faecal DNA. Mol. Ecol. 1998, 7, 1423–1428. [Google Scholar] [CrossRef]
- Copley, T.R.; Giovenazzo, P.; Jabaji, S.H. Detection of Nosema apis and N. ceranae in honeybee bottom scraps and frass in naturally infected hives. Apidologie 2012, 43, 753–760. [Google Scholar] [CrossRef]
- Harpur, B.A.; Dey, A.; Albert, J.R.; Patel, S.; Hines, H.M.; Hasselmann, M.; Packer, L.; Zayed, A. Queens and Workers Contribute Differently to Adaptive Evolution in Bumble Bees and Honey Bees. Genome Biol. Evol. 2017, 9, 2395–2402. [Google Scholar] [CrossRef]
- Kapheim, K.M.; Pan, H.; Li, C.; Blatti, C.; Harpur, B.A.; Ioannidis, P.; Jones, B.M.; Kent, C.F.; Ruzzante, L.; Sloofman, L.; et al. Draft Genome Assembly and Population Genetics of an Agricultural Pollinator, the Solitary Alkali Bee (Halictidae: Nomia melanderi). G3 GenesGenomesGenet. 2019, 9, 625–634. [Google Scholar] [CrossRef]
- Françoso, E.; Araujo, N.D.; Ricardo, P.C.; Santos, P.K.F.; Zuntini, A.R.; Arias, M.C. Evolutionary perspectives on bee mtDNA from mito-OMICS analyses of a solitary species. Apidologie 2020, 51, 531–544. [Google Scholar] [CrossRef]
- Lindauer, M.; Watkin, M.B. Division of Labour in the Honeybee Colony. Bee World 1953, 34, 63–73. [Google Scholar] [CrossRef]
- Koeniger, G.; Koeniger, N.; Tiesler, F.-K. Paarungsbiologie und Paarungskontrolle bei der Honigbiene, 1st ed.; Druck H. Buschhausen: Herten, Germany, 2014. [Google Scholar]
- Free, J.B.; Williams, I.H. Factors determining the rearing and rejection of drones by the honeybee colony. Anim. Behav. 1975, 23, 650–675. [Google Scholar] [CrossRef]
- Brascamp, E.W.; Vanders, T.H.V.; Vientjes, Y.C.J.; Bijma, P. Prospects for genomic selection in honey-bee breeding. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018; Volume 11, pp. 1–7. [Google Scholar]
- Moškrič, A.; Mole, K.; Prešern, J. EPIC markers of the genus Apis as diagnostic tools for detection of honey fraud. Food Control 2020, 107634. [Google Scholar] [CrossRef]
- Soares, S.; Grazina, L.; Mafra, I.; Costa, J.; Pinto, M.A.; Oliveira, M.B.P.; Amaral, J.S. Towards honey authentication: Differentiation of Apis mellifera subspecies in European honeys based on mitochondrial DNA markers. Food Chem. 2019, 283, 294–301. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | ID | Chr. Num | Location | dCAPS Oligonucleotide Primer Sequence (Direction 5′ to 3′) | Restriction Endonuclease |
|---|---|---|---|---|---|
| SNP2 | AMB-00457689 | 3 | 10425353 | SNP2F: CCGTGTTCCTTCCTCTCTTTCTCAGC SNP2R: CGAGTTCTCGTCCAGGCATC | AluI |
| SNP3 | AMB-00745078 | 6 | 1398456 | SNP3F: TCAACCTTCTTTCCTTCTTCCT SNP3R: CAAAACCCCATAAACGCCCC | MnlI |
| SNP2 | SNP3 | |
|---|---|---|
| Allele | T/C | C/T |
| annealing T (°C) | 55 | 50 |
| fragment length (bp) | 217 | 201 |
| restricted fragments size (bp) | 192/25 | 172/29 |
| Extracted DNA Source | Mean Quantity (ng/μL) (Range) | Mean 260:280 OD Ratio (Range) |
|---|---|---|
| Exuviae | 2.7 (0.03 to 21.5) | 1.62 (0.35 to 2.13) |
| Feces | 4.5 (0.8 to 21.3) | 1.45 (1.09 to 1.79) |
| Wing clippings | 0.91 (0.2 to 2.39) | 1.19 (1.01 to 1.88) |
| Legs | 38.72 (7.11 to 239) | 2.01 (1.72 to 2.18) |
| Antennae | 0.5 (0.12 to 1) | 1.35 (1.06 to 1.56) |
| DNA Fragment | Exuviae +/Total | Feces +/Total | Wing-Clippings +/Total | Legs +/Total | Antennae +/Total |
|---|---|---|---|---|---|
| tRNAleu-COX2 | 54/95 | 16/16 | 6/8 | 41/41 | 3/4 |
| COI | 38/53 | 15/16 | 8/8 | 26/27 | 2/4 |
| SNP2 | 29/36 | 15/16 | 5/5 | 40/40 | n.a. |
| SNP3 | 27/36 | 15/16 | 5/5 | 39/39 | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bubnič, J.; Mole, K.; Prešern, J.; Moškrič, A. Non-Destructive Genotyping of Honeybee Queens to Support Selection and Breeding. Insects 2020, 11, 896. https://doi.org/10.3390/insects11120896
Bubnič J, Mole K, Prešern J, Moškrič A. Non-Destructive Genotyping of Honeybee Queens to Support Selection and Breeding. Insects. 2020; 11(12):896. https://doi.org/10.3390/insects11120896
Chicago/Turabian StyleBubnič, Jernej, Katarina Mole, Janez Prešern, and Ajda Moškrič. 2020. "Non-Destructive Genotyping of Honeybee Queens to Support Selection and Breeding" Insects 11, no. 12: 896. https://doi.org/10.3390/insects11120896
APA StyleBubnič, J., Mole, K., Prešern, J., & Moškrič, A. (2020). Non-Destructive Genotyping of Honeybee Queens to Support Selection and Breeding. Insects, 11(12), 896. https://doi.org/10.3390/insects11120896