Miocene Diversification and High-Altitude Adaptation of Parnassius Butterflies (Lepidoptera: Papilionidae) in Qinghai–Tibet Plateau Revealed by Large-Scale Transcriptomic Data

 ,
,  ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. mRNA-Seq Library Construction, Illumina Sequencing, Assembly, and Annotation
2.3. Check for Cross-Contaminations
2.4. Ortholog Identification and Alignment
2.5. Phylogenetic Analysis
2.6. Divergence Time Estimation
2.7. Evolutionary Rate and Positive Selection Analyses
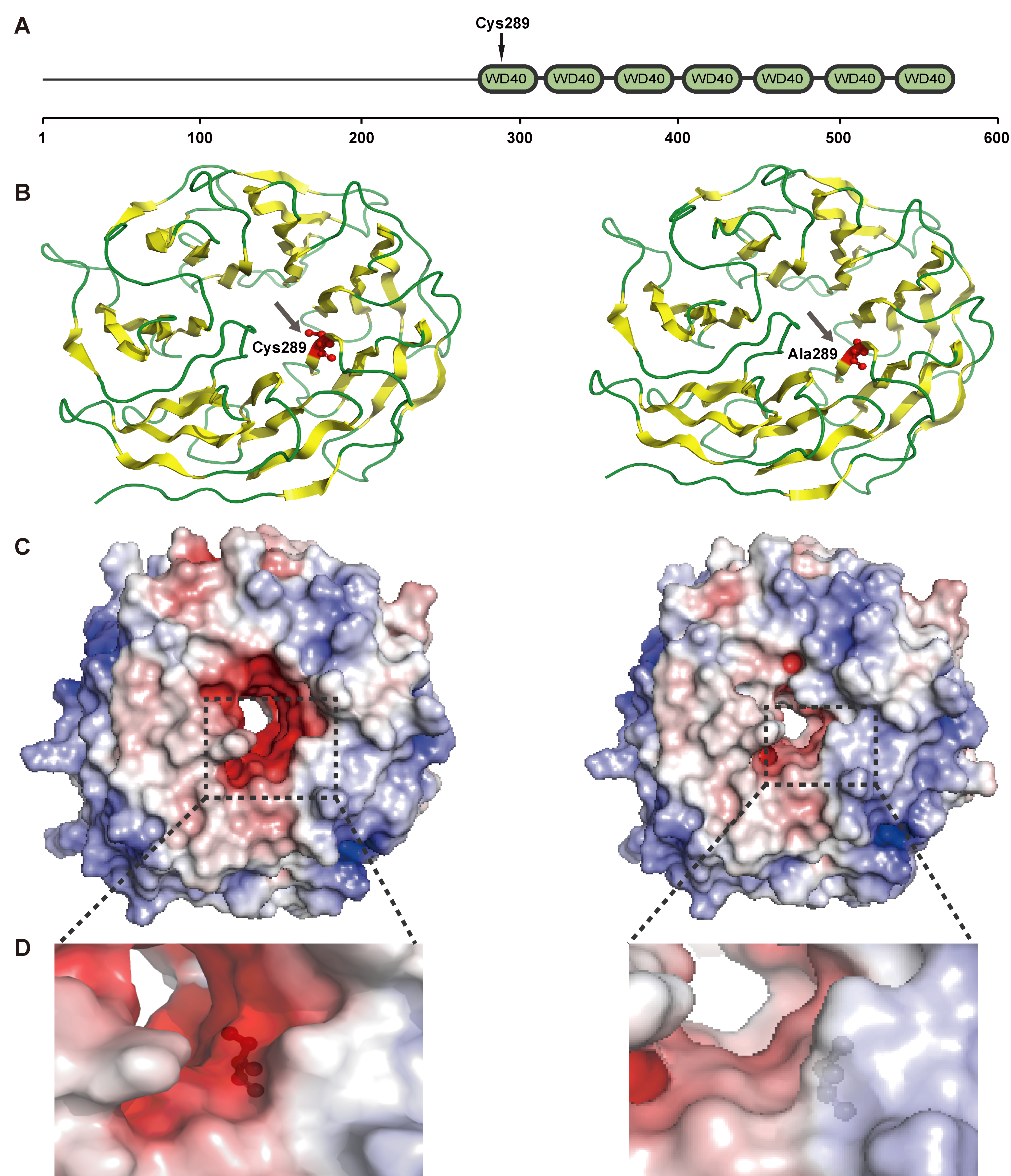
2.8. Protein Three-Dimensional (3D) Structure Simulation
3. Results
3.1. Sequencing, De Novo Assembly, and Annotation
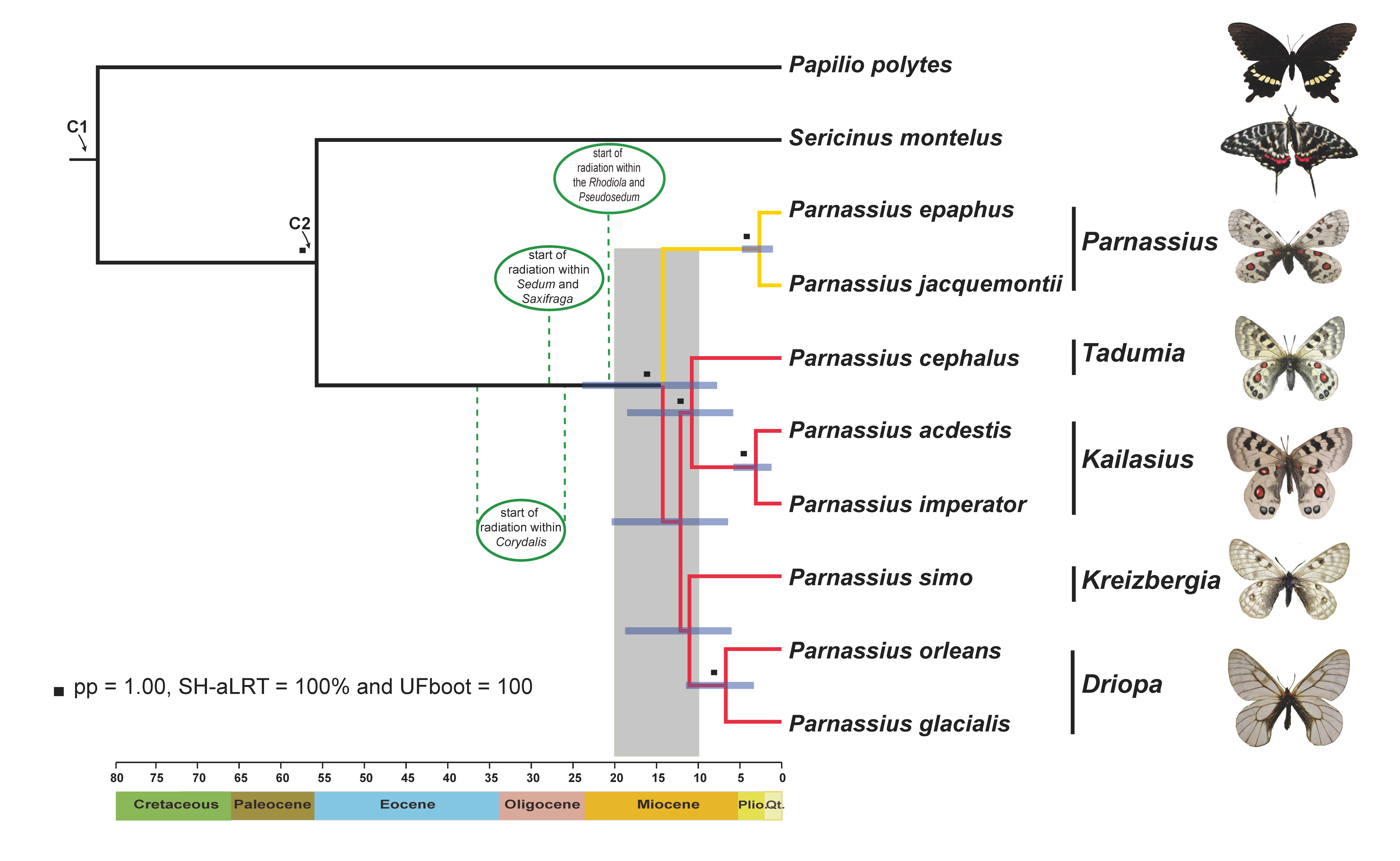
3.2. Phylogenetic Analysis
3.3. Divergence Time Estimates
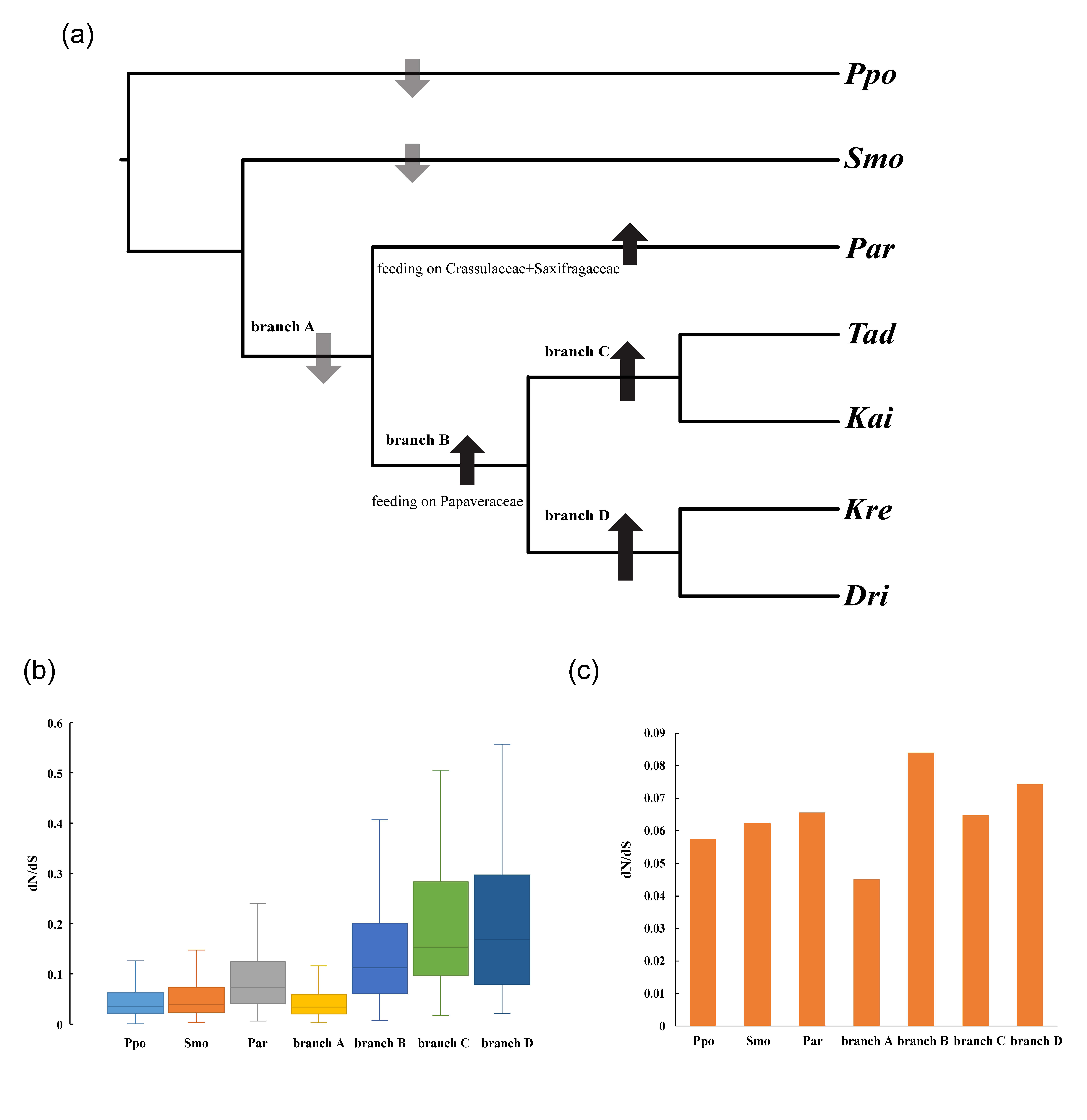
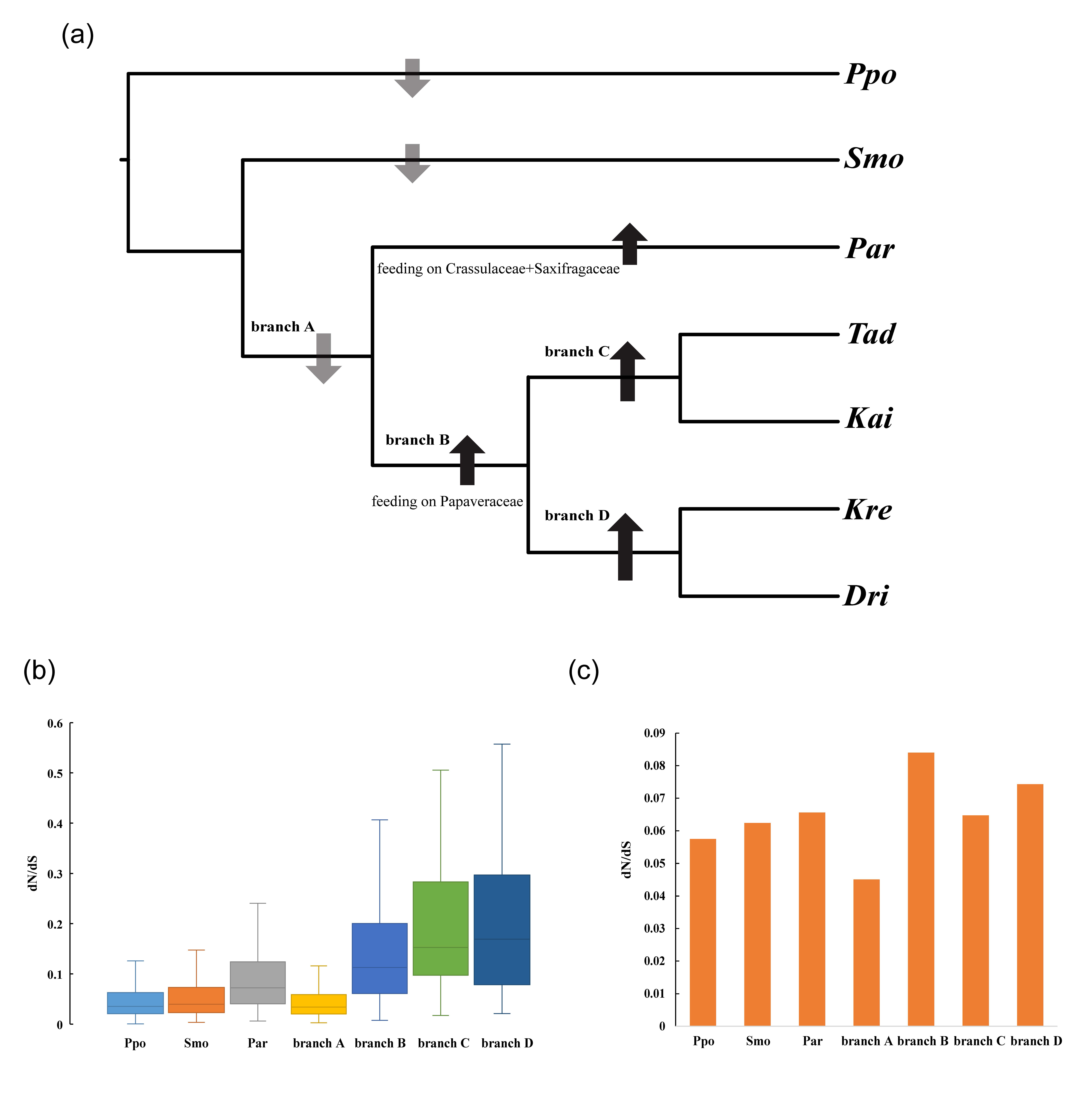
3.4. Accelerated Evolution of the Subgeneric Diversification
3.5. Positively Selected Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ding, D.; Liu, G.J.; Hou, L.; Gui, W.Y.; Chen, B.; Kang, L. Genetic variation in PTPN1 contributes to metabolic adaptation to high-altitude hypoxia in Tibetan migratory locusts. Nat. Commun. 2018, 9, 4991. [Google Scholar] [CrossRef]
- Sun, Y.B.; Fu, T.T.; Jin, J.Q.; Murphy, R.W.; Hillis, D.M.; Zhang, Y.P.; Che, J. Species groups distributed across elevational gradients reveal convergent and continuous genetic adaptation to high elevations. Proc. Natl. Acad. Sci. USA 2018, 115, E10634–E10641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Zhang, J.Q.; Nie, Z.L.; Zhong, Y.; Sun, H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front. Genet. 2014, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.Z.; Wang, L.Z.; Han, J.; Tang, X.L.; Ma, M.; Wang, K.; Zhang, X.; Ren, Q.; Chen, Q.; Qiu, Q. Comparative transcriptomic analysis revealed adaptation mechanism of Phrynocephalus erythrurus, the highest altitude Lizard living in the Qinghai-Tibet Plateau. BMC Evol. Biol. 2015, 15, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.M.; Mao, L.F.; Yang, T.; Ye, J.F.; Liu, B.; Li, H.L.; Sun, M.; Miller, J.T.; Mathews, S.; Hu, H.H.; et al. Evolutionary history of the angiosperm flora of China. Nature 2018, 554, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Leneveu, J.; Chichvarkhin, A.; Wahlberg, N. Varying rates of diversification in the genus Melitaea (Lepidoptera: Nymphalidae) during the past 20 million years. Biol. J. Linn. Soc. 2009, 97, 346–361. [Google Scholar] [CrossRef] [Green Version]
- Päckert, M.; Martens, J.; Sun, Y.H.; Tietze, D.T. Evolutionary history of passerine birds (Aves: Passeriformes) from the Qinghai-Tibetan plateau: From a pre-Quarternary perspective to an integrative biodiversity assessment. J. Ornithol. 2015, 156, S355–S365. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, S. Extinction vs. Rapid radiation: The juxtaposed evolutionary histories of coelotine spiders support the Eocene–Oligocene orogenesis of the Tibetan Plateau. Syst. Biol. 2017, 66, 988–1006. [Google Scholar] [CrossRef]
- Zhao, D.J.; Zhang, Z.Y.; Cease, A.; Harrison, J.; Kang, L. Efficient utilization of aerobic metabolism helps Tibetan locusts conquer hypoxia. BMC Genom. 2013, 14, 631. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Haddad, G.G. Genetic analysis of hypoxia tolerance and susceptibility in Drosophila and humans. Annu. Rev. Genom. Hum. Genet. 2013, 14, 25–43. [Google Scholar] [CrossRef]
- Cui, M.M.; Hu, P.; Wang, T.; Tao, J.; Zong, S. Differential transcriptome analysis reveals genes related to cold tolerance in seabuckthorn carpenter moth, Eogystia hippophaecolus. PLoS ONE 2017, 12, e0187105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazari, V.; Zakharov, E.V.; Sperling, F.A. Phylogeny, historical biogeography, and taxonomic ranking of Parnassiinae (Lepidoptera, Papilionidae) based on morphology and seven genes. Mol. Phylogenet. Evol. 2007, 42, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Michel, F.; Rebourg, C.; Cosson, E.; Descimon, H. Molecular phylogeny of Parnassiinae butterflies (Lepidoptera: Papilionidae) based on the sequences of four mitochondrial DNA segments. Ann. Soc. Entomol. Fr. 2008, 44, 1–36. [Google Scholar] [CrossRef]
- Condamine, F.L.; Rolland, J.; Höhna, S.; Sperling, F.A.; Sanmartín, I. Testing the role of the Red Queen and Court Jester as drivers of the macroevolution of Apollo butterflies. Syst. Biol. 2018, 67, 940–964. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, Y.; Xia, C.; Huang, D.; Cao, Y.; Hao, J.; Zhu, C. The complete mitochondrial genome of Parnassius actius (Lepidoptera: Papilionidae: Parnassinae) with the related phylogenetic analysis. Zool. Syst. 2018, 43, 1–17. [Google Scholar] [CrossRef]
- Wilhelm, B.T.; Landry, J.R. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257. [Google Scholar] [CrossRef]
- Hu, J.W.; You, F.; Wang, Q.; Weng, S.D.; Liu, H.; Wang, L.J.; Zhang, P.J.; Tan, X.G. Transcriptional responses of olive flounder (Paralichthys olivaceus) to low temperature. PLoS ONE 2014, 9, e108582. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Simion, P.; Belkhir, K.; Francois, C.; Veyssier, J.; Rink, J.C.; Manuel, M.; Philippe, H.; Telford, M.J. A software tool ‘CroCo’ detects pervasive cross-species contamination in next generation sequencing data. BMC Biol. 2018, 16, 28. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Chazot, N.; Wahlberg, N.; Freitas, A.V.L.; Mitter, C.; Labandeira, C.; Sohn, J.C.; Sahoo, R.K.; Seraphim, N.; de Jong, R.; Heikkila, M. Priors and posteriors in Bayesian timing of divergence analyses: The age of butterflies revisited. Syst. Biol. 2019, 68, 797–813. [Google Scholar] [CrossRef]
- Allio, R.; Scornavacca, C.; Nabholz, B.; Clamens, A.L.; Sperling, F.A.; Condamine, F.L. Whole genome shotgun phylogenomics resolves the pattern and timing of swallowtail butterfly evolution. Syst. Biol. 2020, 69, 38–60. [Google Scholar] [CrossRef]
- Durden, C.J.; Rose, H. Butterflies from the Middle Eocene: The Earliest Occurrence of Fossil Papilionoidea (Lepidoptera); Texas Memorial Museum, The University of Texas at Austin: Austin, TX, USA, 1978. [Google Scholar]
- Scudder, S.H. Fossil butterflies. Mem. Am. Assoc. Adv. Sci. 1875, 1, 1–99. [Google Scholar]
- Drummond, A.J.; Bouckaert, R.R. Bayesian Evolutionary Analysis with BEAST; Cambridge University Press: London, UK, 2015; pp. 98–125. [Google Scholar]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 20 December 2019).
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, Y.; Zhang, Z.; He, S. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus. Genome Biol. Evol. 2014, 7, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yang, L.; Zhou, K.; Zhang, Y.; Song, Z.; He, S. Evidence for adaptation to the Tibetan Plateau inferred from Tibetan loach transcriptomes. Genome Biol. Evol. 2015, 7, 2970–2982. [Google Scholar] [CrossRef] [Green Version]
- Goodman, M.; Sterner, K.N.; Islam, M.; Uddin, M.; Sherwood, C.C.; Hof, P.R.; Hou, Z.C.; Lipovich, L.; Jia, H.; Grossman, L.I. Phylogenomic analyses reveal convergent patterns of adaptive evolution in elephant and human ancestries. Proc. Natl. Acad. Sci. USA 2009, 106, 20824–20829. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Z.; Nielsen, R.; Yang, Z.H. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. Ccp4 Newslett. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Unni, S.; Huang, Y.; Hanson, R.M.; Tobias, M.; Krishnan, S.; Li, W.W.; Nielsen, J.E.; Baker, N.A. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. Comput. Chem. 2011, 32, 1488–1491. [Google Scholar] [CrossRef] [Green Version]
- Theissinger, K.; Falckenhayn, C.; Blande, D.; Toljamo, A.; Gutekunst, J.; Makkonen, J.; Jussila, J.; Lyko, F.; Schrimpf, A.; Schulz, R.; et al. De Novo assembly and annotation of the freshwater crayfish Astacus astacus transcriptome. Mar. Genom. 2016, 28, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Condamine, F.L.; Sperling, F.A.H.; Kergoat, G.J. Global biogeographical pattern of swallowtail diversification demonstrates alternative colonization routes in the Northern and Southern hemispheres. J. Biogeogr. 2013, 40, 9–23. [Google Scholar] [CrossRef]
- Guo, Z.T.; Ruddiman, W.F.; Hao, Q.Z.; Wu, H.B.; Qiao, Y.S.; Zhu, R.X.; Peng, S.Z.; Wei, J.J.; Yuan, B.Y.; Liu, T.S. Onset of Asian desertification by 22 Myr ago inferred from loess deposits in China. Nature 2002, 416, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Zhao, X.X.; Liu, Z.F.; Lippert, P.C.; Graham, S.A.; Coe, R.S.; Yi, H.; Zhu, L.; Liu, S.; Li, Y. Constraints on the early uplift history of the Tibetan Plateau. Proc. Natl. Acad. Sci. USA 2008, 105, 4987–4992. [Google Scholar] [CrossRef] [Green Version]
- Favre, A.; Packert, M.; Pauls, S.U.; Jahnig, S.C.; Uhl, D.; Michalak, I.; Muellner-Riehl, A.N. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. 2015, 90, 236–253. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Meng, S.Y.; Allen, G.A.; Wen, J.; Rao, G.Y. Rapid radiation and dispersal out of the Qinghai-Tibetan Plateau of an alpine plant lineage Rhodiola (Crassulaceae). Mol. Phylogenet. Evol. 2014, 77, 147–158. [Google Scholar] [CrossRef]
- Pérez-Gutiérrez, M.A.; Romero-García, A.T.; Fernández, M.C.; Blanca, G.; Salinas-Bonillo, M.J.; Suárez-Santiago, V.N. Evolutionary history of fumitories (subfamily Fumarioideae, Papaveraceae): An old story shaped by the main geological and climatic events in the Northern Hemisphere. Mol. Phylogenet. Evol. 2015, 88, 75–92. [Google Scholar] [CrossRef]
- Ebersbach, J.; Muellner-Riehl, A.N.; Michalak, I.; Tkach, N.; Hoffmann, M.H.; Roser, M.; Sun, H.; Favre, A. In and out of the Qinghai-Tibet Plateau: Divergence time estimation and historical biogeography of the large arctic-alpine genus Saxifraga L. J. Biogeogr. 2016, 44, 900–910. [Google Scholar] [CrossRef]
- Omoto, K.; Yonezawa, T.; Shinkawa, T. Molecular systematics and evolution of the recently discovered “Parnassian” butterfly (Parnassius davydovi Churkin, 2006) and its allied species (Lepidoptera, Papilionidae). Gene 2009, 441, 80–88. [Google Scholar] [CrossRef]
- Marchesini, M.; Ogoti, Y.; Fiorini, E.; Samur, A.A.; Nezi, L.; D’Anca, M.; Storti, P.; Samur, M.K.; Ganan-Gomez, I.; Fulciniti, M.T.; et al. ILF2 is a regulator of RNA splicing and DNA damage response in 1q21-amplified multiple myeloma. Cancer Cell 2017, 32, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Wolkowicz, U.M.; Cook, A.G. NF45 dimerizes with NF90, Zfr and SPNR via a conserved domain that has a nucleotidyltransferase fold. Nucleic Acids Res. 2012, 40, 9356–9368. [Google Scholar] [CrossRef] [Green Version]
- Dahan, O.; Kupiec, M. The Saccharomyces cerevisiae gene CDC40/PRP17 controls cell cycle progression through splicing of the ANC1 gene. Nucleic Acids Res. 2004, 32, 2529–2540. [Google Scholar] [CrossRef] [Green Version]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Roberts, R. WD-repeat proteins: Structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 2001, 58, 2085–2097. [Google Scholar] [CrossRef]
- Gorfinkiel, N.; Sierra, J.; Callejo, A.; Ibanez, C.; Guerrero, I. The Drosophila ortholog of the human Wnt inhibitor factor Shifted controls the diffusion of lipid-modified Hedgehog. Dev. Cell 2005, 8, 241–253. [Google Scholar] [CrossRef]
- Glise, B.; Miller, C.A.; Crozatier, M.; Halbisen, M.A.; Wise, S.; Olson, D.J.; Vincent, A.; Blair, S.S. Shifted, the Drosophila ortholog of Wnt inhibitory factor-1, controls the distribution and movement of Hedgehog. Dev. Cell 2005, 8, 255–266. [Google Scholar] [CrossRef]
- Shi, X.X.; Huang, Y.J.; Begum, M.A.; Zhu, M.F.; Li, F.Q.; Zhang, M.J.; Zhou, W.W.; Mao, C.G.; Zhu, Z.R. A neutral ceramidase, NlnCDase, is involved in the stress responses of brown planthopper, Nilaparvata lugens (Stål). Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Acharya, J.K.; Dasgupta, U.; Rawat, S.S.; Yuan, C.Q.; Sanxaridis, P.D.; Yonamine, I.; Karim, P.; Nagashima, K.; Brodsky, M.H.; Tsunoda, S.; et al. Cell-nonautonomous function of ceramidase in photoreceptor homeostasis. Neuron 2008, 57, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Okino, N.; Kakuta, Y.; Hijikata, A.; Okano, H.; Goda, H.M.; Tani, M.; Sueyoshi, N.; Kambayashi, K.; Matsumura, H.; et al. Mechanistic insights into the hydrolysis and synthesis of ceramide by neutral ceramidase. J. Biol. Chem. 2009, 284, 9566–9577. [Google Scholar] [CrossRef] [Green Version]
- Maliekal, P.; Sokolova, T.; Vertommen, D.; Veiga-Da-Cunha, M.; Van Schaftingen, E. Molecular identification of mammalian phosphopentomutase and glucose-1,6-bisphosphate synthase, two members of the alpha-D-phosphohexomutase family. J. Biol. Chem. 2007, 282, 31844–31851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beamer, L.J. Mutations in hereditary phosphoglucomutase 1 deficiency map to key regions of enzyme structure and function. J. Inherit. Metab. Dis. 2015, 38, 243–256. [Google Scholar] [CrossRef]
- Muenks, A.G.; Stiers, K.M.; Beamer, L.J. Sequence-structure relationships, expression profiles, and disease-associated mutations in the paralogs of phosphoglucomutase 1. PLoS ONE 2017, 12, e0183563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.N.; Ree, R.H.; Spicer, R.A.; Xing, Y.W. Ancient orogenic and monsoon-driven assembly of the world’s richest temperate alpine flora. Science 2020, 369, 578–581. [Google Scholar] [CrossRef]
- Despland, E. Butterflies of the high-altitude Atacama Desert: Habitat use and conservation. Front. Genet. 2014, 5, 334. [Google Scholar] [CrossRef] [Green Version]
- Wheat, C.W.; Vogel, H.; Wittstock, U.; Braby, M.F.; Underwood, D.; Mitchell-Olds, T. The genetic basis of a plant-insect coevolutionary key innovation. Proc. Natl. Acad. Sci. USA 2007, 104, 20427–20431. [Google Scholar] [CrossRef] [Green Version]
- Fordyce, J.A. Host shifts and evolutionary radiations of butterflies. Proc. R. Soc. B Biol. Sci. 2010, 277, 3735–3743. [Google Scholar] [CrossRef]
- Su, C.Y.; Shi, Q.H.; Sun, X.Y.; Ma, J.Y.; Li, C.X.; Hao, J.S.; Yang, Q. Dated phylogeny and dispersal history of the butterfly subfamily Nymphalinae (Lepidoptera: Nymphalidae). Sci. Rep. 2017, 7, 8799. [Google Scholar] [CrossRef]
- Peña, C.; Witthauer, H.; Klečková, I.; Fric, Z.; Wahlberg, N. Adaptive radiations in butterflies: Evolutionary history of the genus Erebia (Nymphalidae: Satyrinae). Biol. J. Linn. Soc. 2015, 116, 449–467. [Google Scholar] [CrossRef] [Green Version]
- Drovetski, S.V.; Semenov, G.; Drovetskaya, S.S.; Fadeev, I.V.; Red’kin, Y.A.; Voelker, G. Geographic mode of speciation in a mountain specialist Avian family endemic to the Palearctic. Ecol. Evol. 2013, 3, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
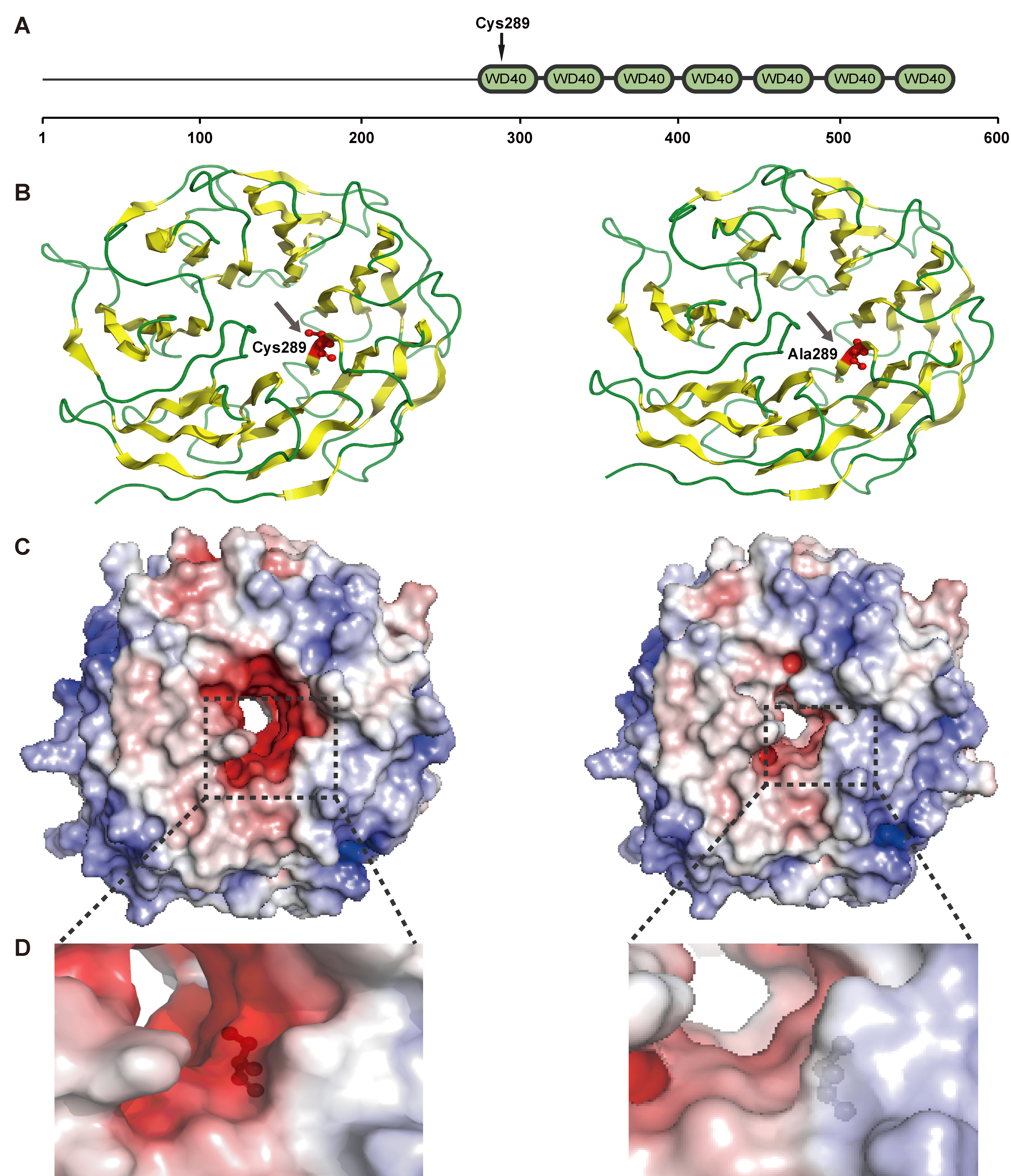{kind=link}
| Major Lineage/Split | Amino Acid Dataset | Nucleotide Dataset | ||
|---|---|---|---|---|
| Mean Age | 95% CI | Mean Age | 95% CI | |
| Genus Parnassius | 14.3 | 7.7–24.0 | 13.0 | 6.8–22.3 |
| Tadumia + Kailasius/Kreizbergia + Driopa | 12.1 | 6.4–20.4 | 11.2 | 5.9–19.3 |
| Tadumia/Kailasius | 10.8 | 5.8–18.6 | 9.9 | 5.2–17.1 |
| Kreizbergia/Driopa | 11.1 | 6.0–18.8 | 10.2 | 5.3–17.5 |
| Gene | Model with Positive Selection (Model A) | Null Model (Model A null) | LRT | p-Value | Biological Category | |||
|---|---|---|---|---|---|---|---|---|
| L1 | Parameters | Positively Selected Sites | L0 | Parameters | 2ΔL | |||
| ILF2 | −1335.05 | k = 1.73 | Ser149Gln 0.98 * | −1337.71 | k = 1.71 | 5.32 | 0.02 | RNA splicing and DNA damage response |
| p0 = 0.97 | p0 = 0.95 | |||||||
| ω0 = 0.02 | ω0 = 0.02 | |||||||
| ps = 0.02 | ||||||||
| ωs = 25.86 | ||||||||
| PRPF17 | −4662.37 | k = 1.65 | Ala289Cys 0.98 * | −4664.23 | k = 1.64 | 3.72 | 0.05 | pre-mRNA splicing and mRNA surveillance |
| p0 = 0.97 | p0 = 0.95 | |||||||
| ω0 = 0.03 | ω0 = 0.03 | |||||||
| ps = 0.01 | ||||||||
| ωs = 14.87 | ||||||||
| SHF | −3105.63 | k = 2.04 | Val300Arg 1.00 ** | −3108.38 | k = 2.02 | 5.50 | 0.02 | morphogenesis and patterning |
| p0 = 0.94 | Lys304Arg 0.98 * | p0 = 0.91 | ||||||
| ω0 = 0.02 | Asp314Arg 0.98 * | ω0 = 0.02 | ||||||
| ps = 0.03 | ||||||||
| ωs = 5.34 | ||||||||
| CDASE | −11227.05 | k = 1.95 | Arg307Val 0.97 * | −11229.24 | k = 1.94 | 4.38 | 0.03 | development and stress responses |
| p0 = 0.92 | His605Ala 0.98 * | p0 = 0.91 | ||||||
| ω0 = 0.05 | ω0 = 0.05 | |||||||
| ps = 0.01 | ||||||||
| ωs = 9.96 | ||||||||
| PGM2 | −5402.44 | k = 1.79 | Cys504Thr 1.00 ** | −5406.12 | k = 1.77 | 7.36 | 0.01 | glycogen metabolism and environmental adaptation |
| p0 = 0.88 | p0 = 0.86 | |||||||
| ω0 = 0.06 | ω0 = 0.06 | |||||||
| ps = 0.01 | ||||||||
| ωs = 15.05 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.; Xie, T.; Wang, Y.; Si, C.; Li, L.; Ma, J.; Li, C.; Sun, X.; Hao, J.; Yang, Q. Miocene Diversification and High-Altitude Adaptation of Parnassius Butterflies (Lepidoptera: Papilionidae) in Qinghai–Tibet Plateau Revealed by Large-Scale Transcriptomic Data. Insects 2020, 11, 754. https://doi.org/10.3390/insects11110754
Su C, Xie T, Wang Y, Si C, Li L, Ma J, Li C, Sun X, Hao J, Yang Q. Miocene Diversification and High-Altitude Adaptation of Parnassius Butterflies (Lepidoptera: Papilionidae) in Qinghai–Tibet Plateau Revealed by Large-Scale Transcriptomic Data. Insects. 2020; 11(11):754. https://doi.org/10.3390/insects11110754
Chicago/Turabian StyleSu, Chengyong, Tingting Xie, Yunliang Wang, Chengcai Si, Luyan Li, Junye Ma, Chunxiang Li, Xiaoyan Sun, Jiasheng Hao, and Qun Yang. 2020. "Miocene Diversification and High-Altitude Adaptation of Parnassius Butterflies (Lepidoptera: Papilionidae) in Qinghai–Tibet Plateau Revealed by Large-Scale Transcriptomic Data" Insects 11, no. 11: 754. https://doi.org/10.3390/insects11110754
APA StyleSu, C., Xie, T., Wang, Y., Si, C., Li, L., Ma, J., Li, C., Sun, X., Hao, J., & Yang, Q. (2020). Miocene Diversification and High-Altitude Adaptation of Parnassius Butterflies (Lepidoptera: Papilionidae) in Qinghai–Tibet Plateau Revealed by Large-Scale Transcriptomic Data. Insects, 11(11), 754. https://doi.org/10.3390/insects11110754