Transcriptomic and Metabolomic Responses of Rice Plants to Cnaphalocrocis medinalis Caterpillar Infestation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
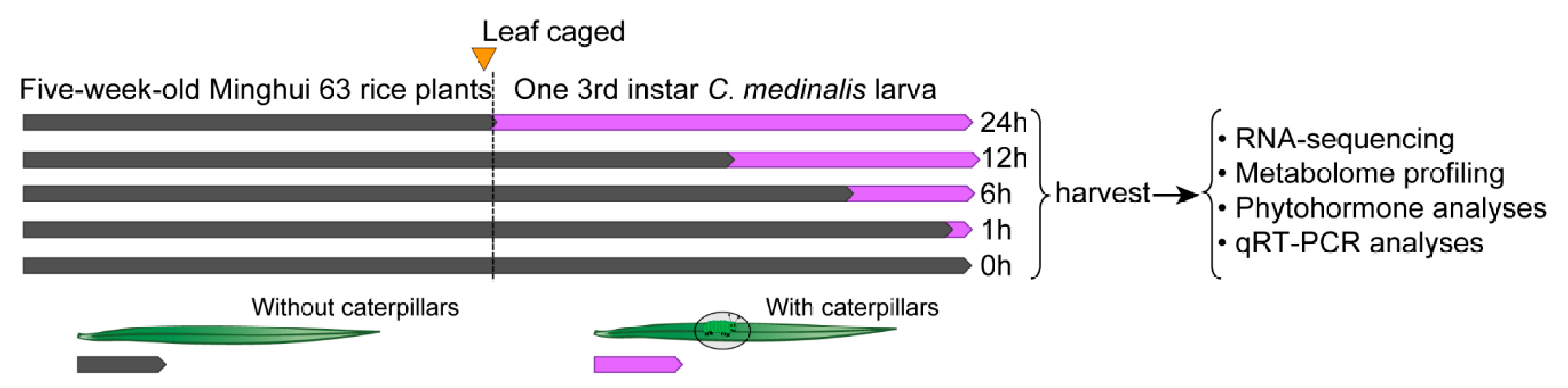
2.1. Plants and Insets
2.2. Sample Preparation
2.3. RNA Isolation, Library Construction, Sequencing, and Validation
2.4. RNA-Sequencing Data Analysis
2.5. Metabolite Profiling
2.6. Metabolite Data Analysis
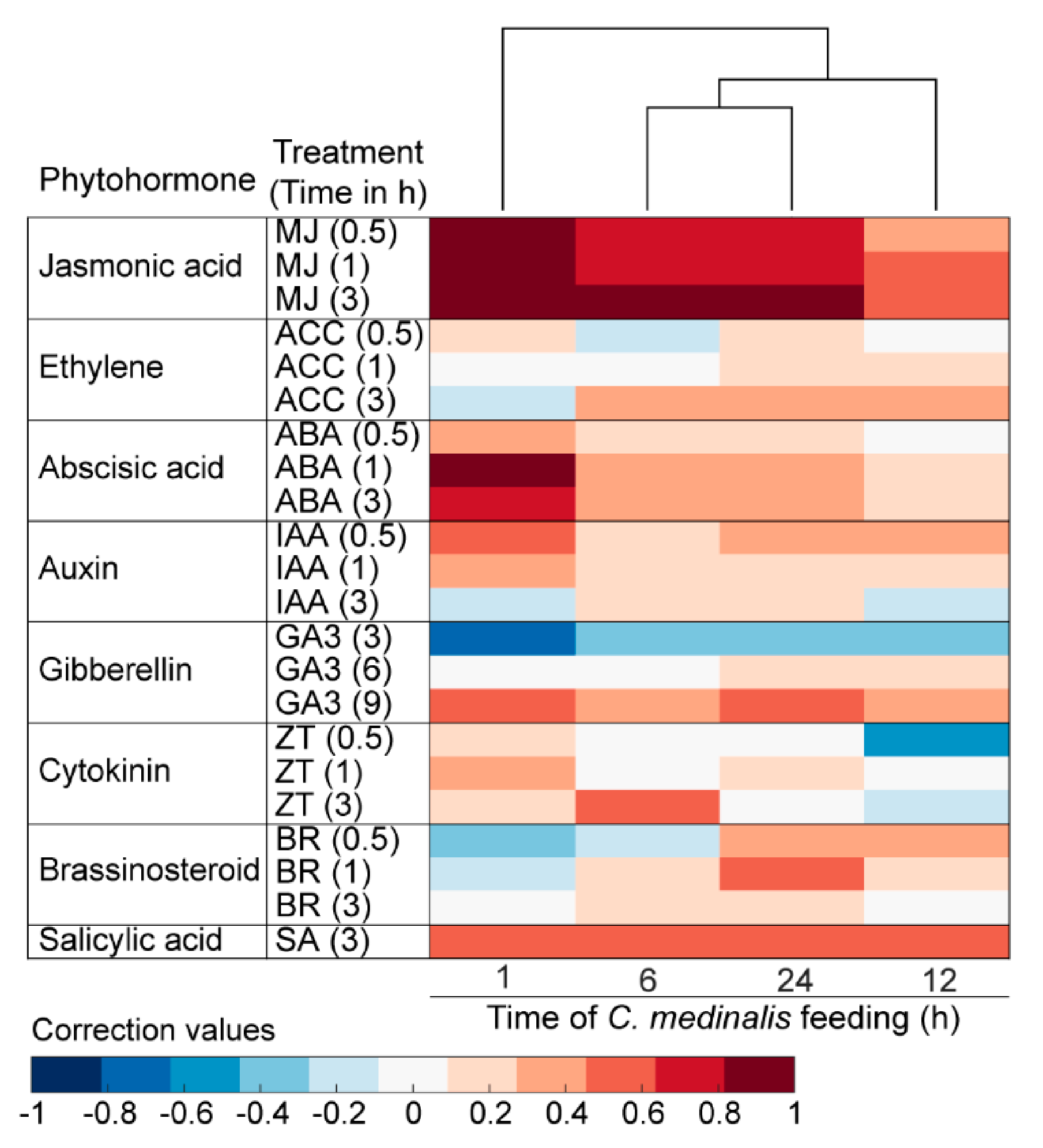
2.7. Phytohormone Analyses
2.7.1. Phytohormone Signatures Analyses
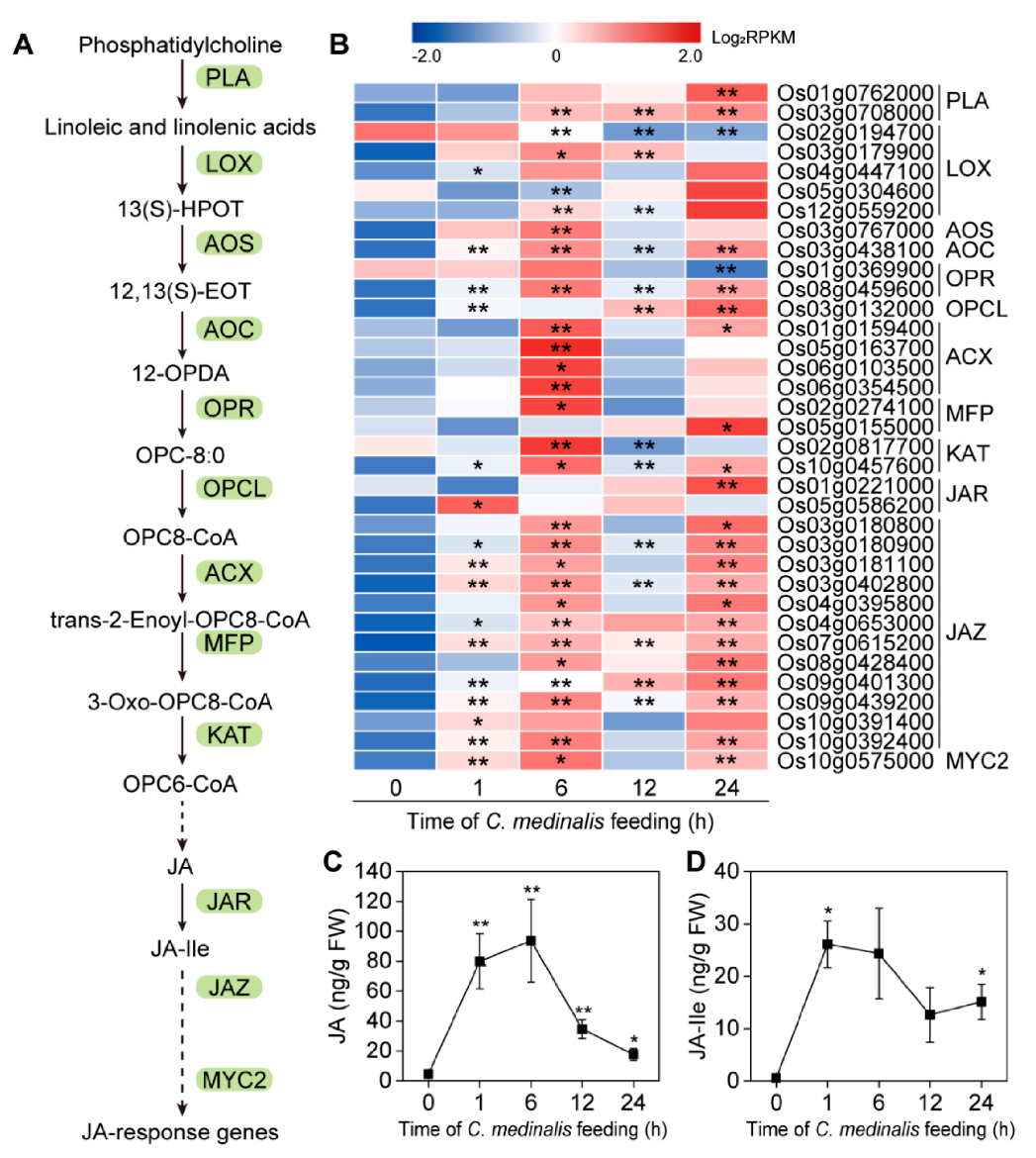
2.7.2. Determination of Endogenous JA and JA-Isoleucine Levels
2.8. Statistical Analysis
2.9. Data Availability
3. Results
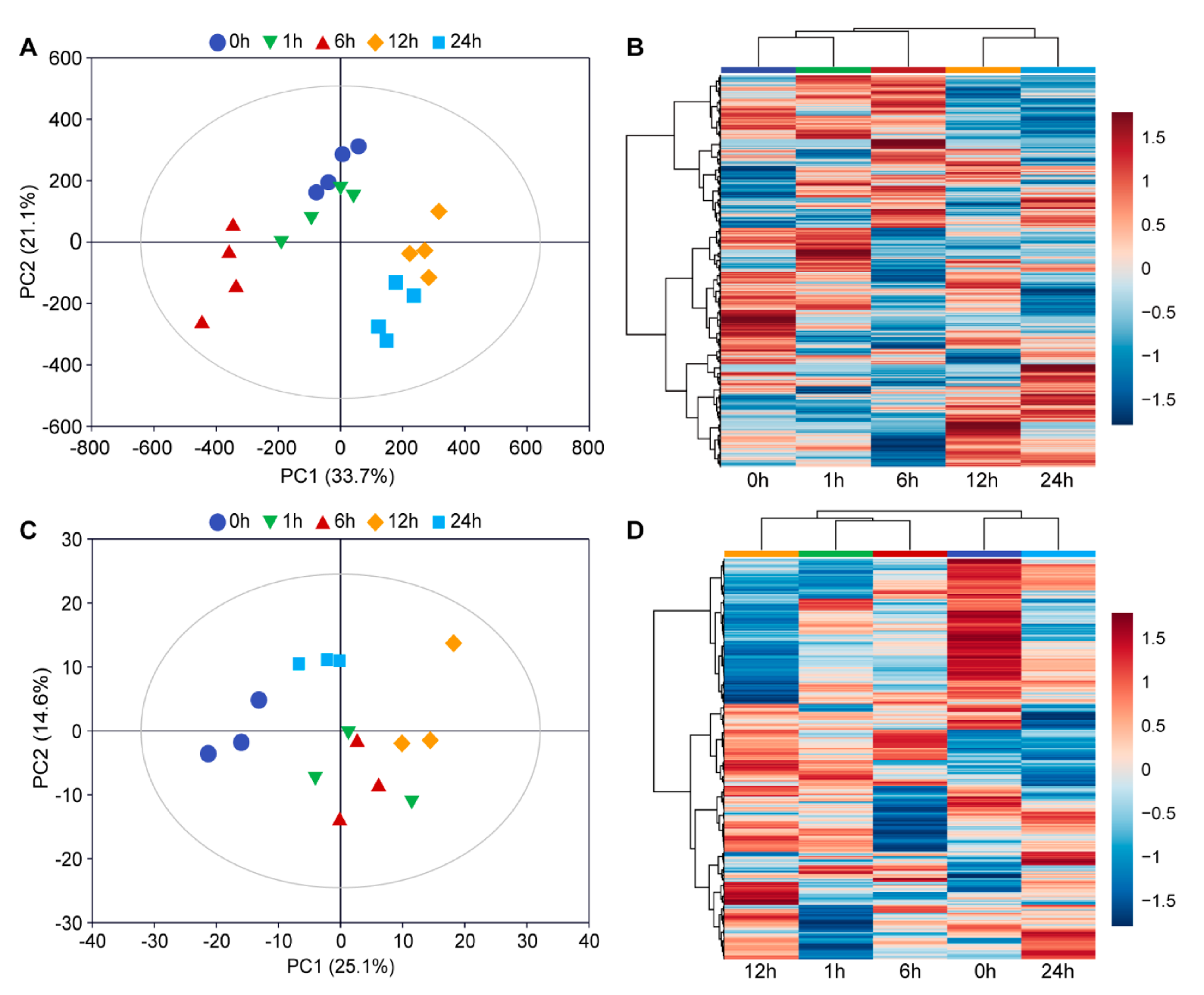
3.1. Overview of Transcriptome and Metabolome Dataset
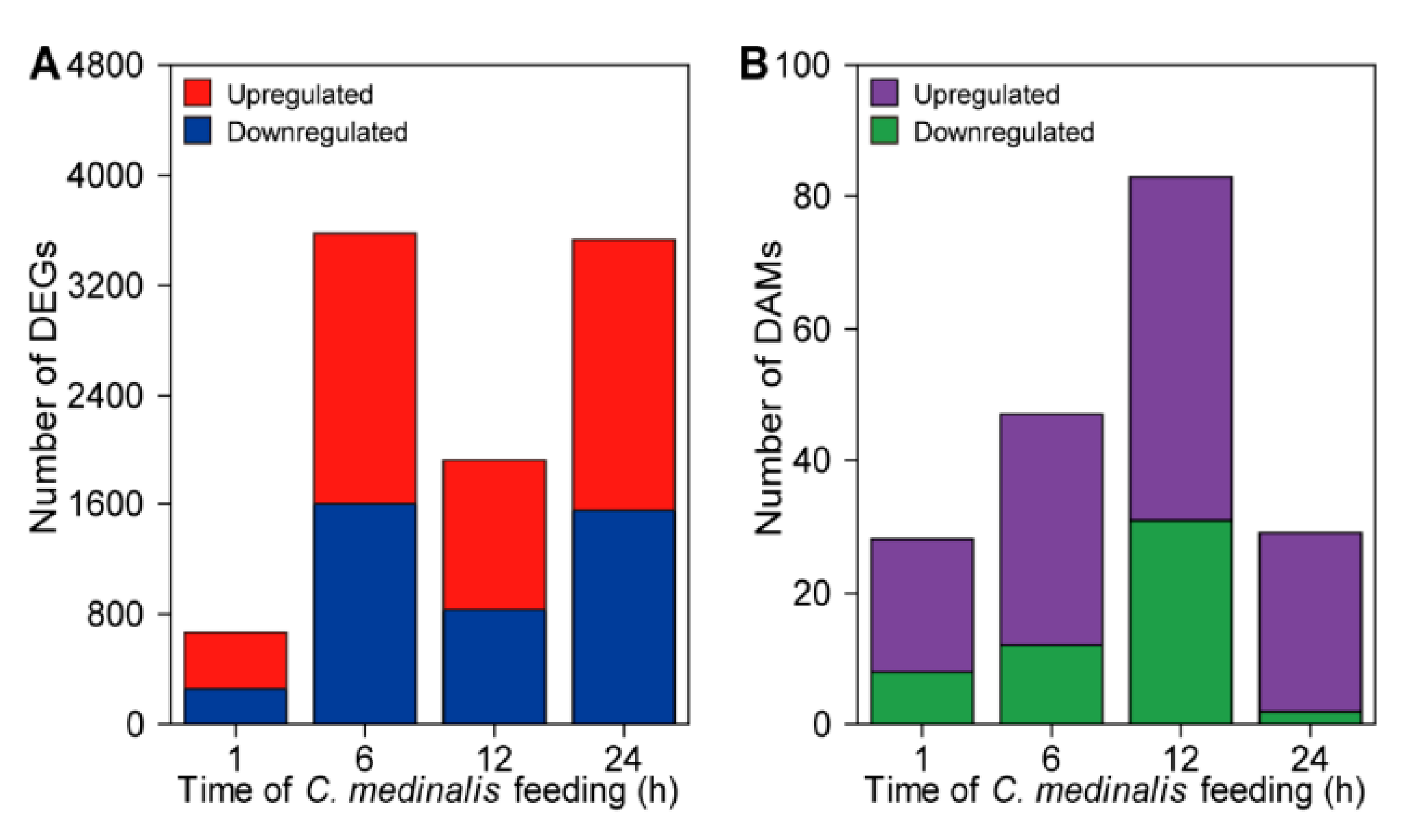
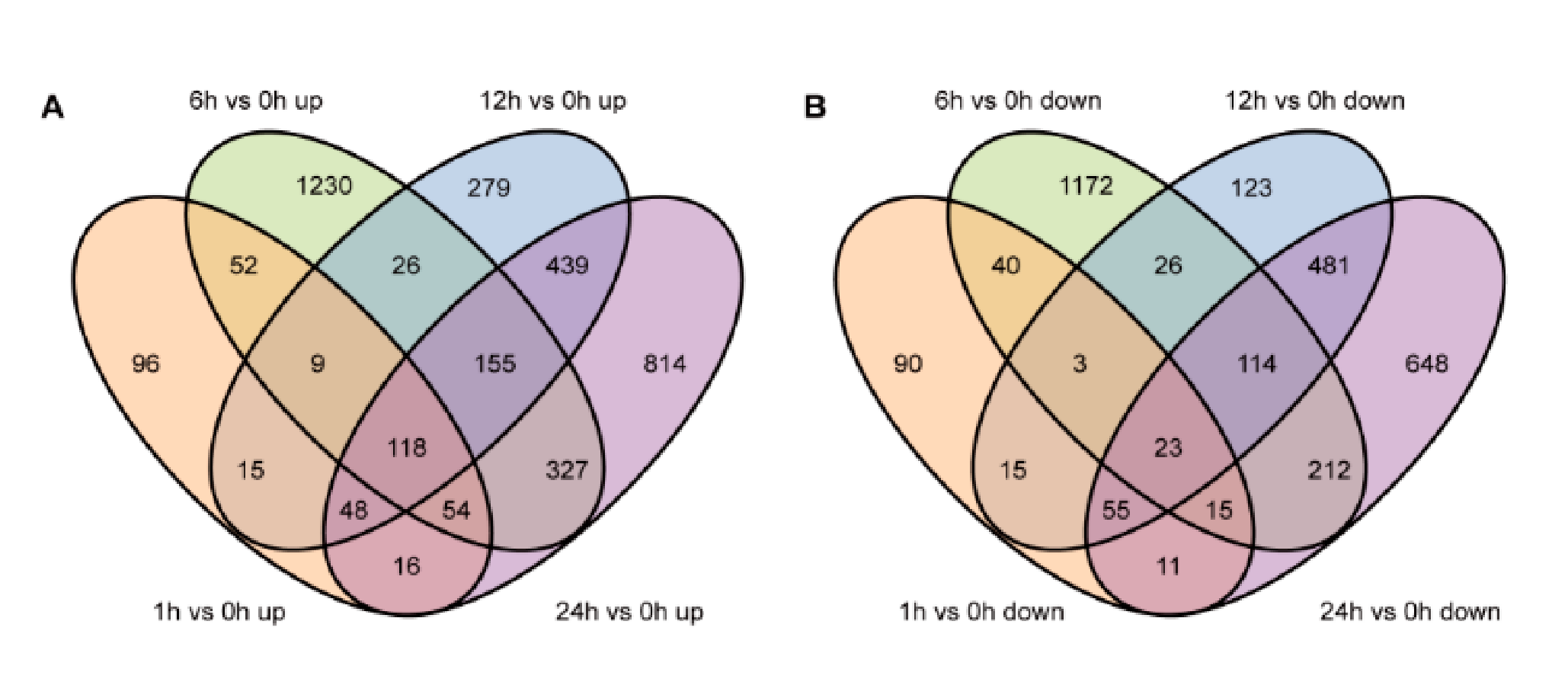
3.2. Analyses of Differentially Expressed Genes and Differentially Accumulated Metabolites
3.3. Identification of Calcium Ions Sensors, Transcription Factors, and Transcriptional Regulators That Responded to C. medinalis Feeding
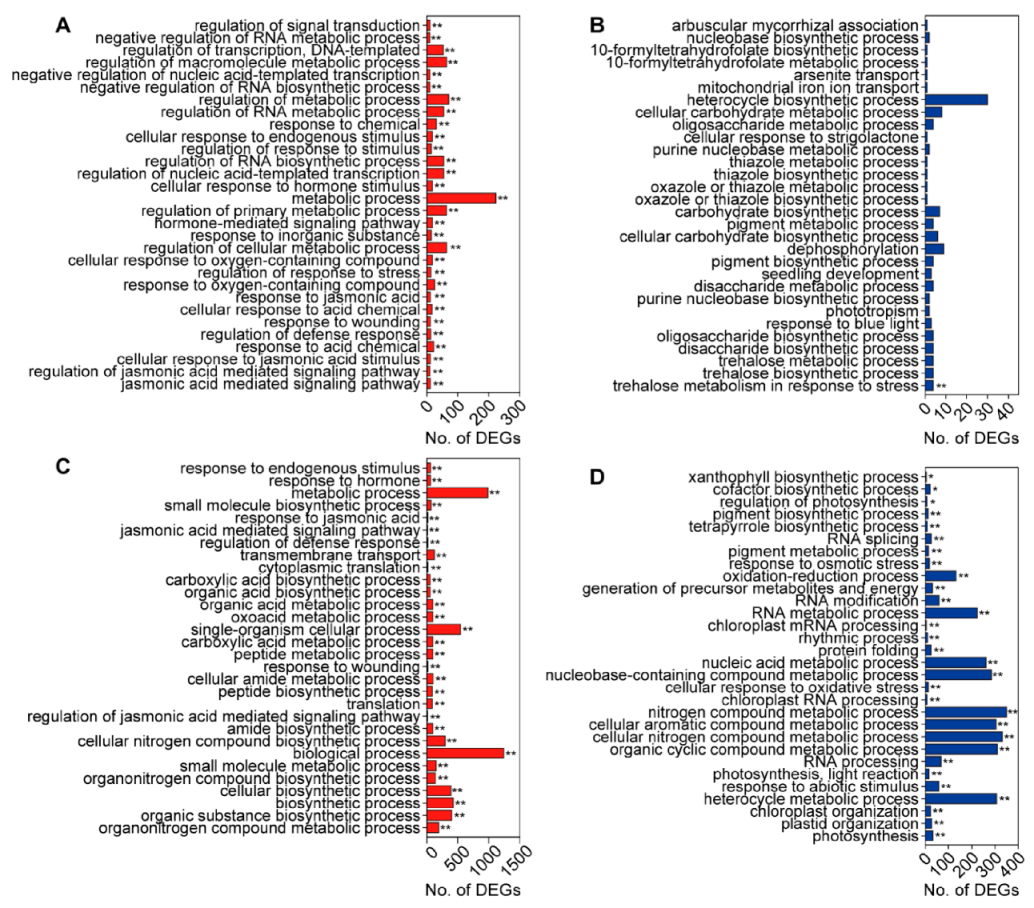
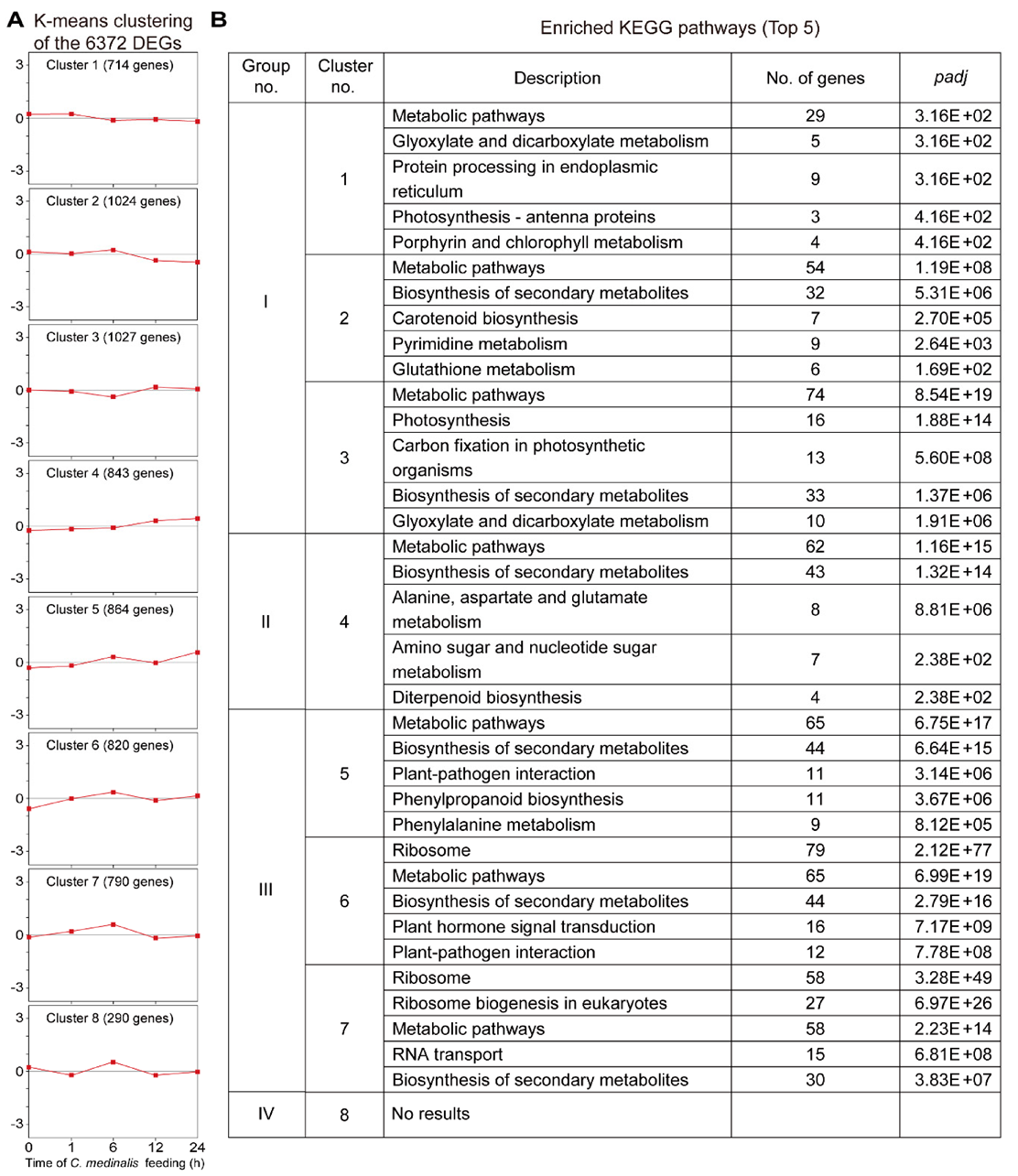
3.4. GO and KEGG Enrichment Analysis of DEGs Induced by C. medinalis Feeding
3.5. Clustering Analyses of the Transcriptome Dataset
3.6. Plant Hormone-Related Genes Induced by C. medinalis Feeding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, J.; Baldwin, I.T. New insights into plant responses to the attack from insect herbivores. Annu. Rev. Genet. 2010, 44, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.A.; Jander, G. Plant immunity to insect herbivores. Annu. Rev. Plant Biol. 2008, 59, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Turlings, T.C.J.; Erb, M. Tritrophic interactions mediated by herbivore-induced plant volatiles: Mechanisms, ecological relevance, and application potential. Annu. Rev. Entomol. 2018, 63, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Stam, J.M.; Kroes, A.; Li, Y.; Gols, R.; van Loon, J.J.; Poelman, E.H.; Dicke, M. Plant interactions with multiple insect herbivores: From community to genes. Annu. Rev. Plant Biol. 2014, 65, 689–713. [Google Scholar] [CrossRef]
- Barah, P.; Bones, A.M. Multidimensional approaches for studying plant defence against insects: From ecology to omics and synthetic biology. J. Exp. Bot. 2015, 66, 479–493. [Google Scholar] [CrossRef]
- Erb, M.; Reymond, P. Molecular interactions between plants and insect herbivores. Annu. Rev. Plant Biol. 2019, 70, 527–557. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, S.; Sun, N.; Liu, H.; Zhao, Y.; Liang, Y.; Zhang, L.; Han, Y. Functional diversity of jasmonates in rice. Rice 2015, 8, 42. [Google Scholar] [CrossRef]
- Li, Y.; Hallerman, E.M.; Wu, K.; Peng, Y. Insect-resistant genetically engineered crops in China: Development, application, and prospects for use. Annu. Rev. Entomol. 2020, 65, 273–292. [Google Scholar] [CrossRef]
- Savary, S.; Willocquet, L.; Elazegui, F.A.; Castilla, N.P.; Teng, P.S. Rice pest constraints intropical Asia: Quantification of yield losses due to rice pests in a range of production situations. Plant Dis. 2000, 84, 357–369. [Google Scholar] [CrossRef]
- Chen, M.; Shelton, A.; Ye, G.Y. Insect-resistant genetically modified rice in China: From research to commercialization. Annu. Rev. Entomol. 2011, 56, 81–101. [Google Scholar] [CrossRef]
- Liu, Q.; Hallerman, M.E.; Peng, Y.; Li, Y. Development of Bt rice and Bt maize in China and their efficacy in target pest control. Int. J. Mol. Sci. 2016, 17, 1561. [Google Scholar] [CrossRef] [PubMed]
- Chintalapati, P.; Balakrishnan, D.; Venu Gopal Nammi, T.V.; Javvaji, S.; Muthusamy, S.K.; Lella Venkata, S.R.; Neelamraju, S.; Katti, G. Phenotyping and genotype × environment interaction of resistance to leaffolder, Cnaphalocrocis medinalis Guenee (Lepidoptera: Pyralidae) in rice. Front. Plant Sci. 2019, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, J.; Horgan, F.G.; Annamalai, S.; Song, K.; Peng, H.; Kang, M.; Win, K.K.; Vongsabouth, T.; Zhang, F.; Tang, R.; et al. Biological control of lepidopteran pests in rice: A multi-nation case study from Asia. J. Integr. Pest Manag. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Senthil-Nathan, S. Effect of methyl jasmonate (MeJA)-induced defenses in rice against the rice leaffolder Cnaphalocrocis medinalis (Guenee) (Lepidoptera: Pyralidae). Pest Manag. Sci. 2019, 75, 460–465. [Google Scholar] [CrossRef]
- Kalaivani, K.; Kalaiselvi, M.M.; Senthil-Nathan, S. Effect of Methyl Salicylate (MeSA) induced changes in rice plant (Oryza sativa) that affect growth and development of the rice leaffolder, Cnaphalocrocis medinalis. Physiol. Mol. Plant Pathol. 2018, 101, 116–126. [Google Scholar] [CrossRef]
- Shi, J.H.; Sun, Z.; Hu, X.J.; Jin, H.; Foba, C.N.; Liu, H.; Wang, C.; Liu, L.; Li, F.F.; Wang, M.Q. Rice defense responses are induced upon leaf rolling by an insect herbivore. BMC Plant Biol. 2019, 19, 514. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, X.; Tzin, V.; Romeis, J.; Peng, Y.; Li, Y. Combined transcriptome and metabolome analyses to understand the dynamic responses of rice plants to attack by the rice stem borer Chilo suppressalis (Lepidoptera: Crambidae). BMC Plant Biol. 2016, 16, 259. [Google Scholar] [CrossRef]
- Zhang, K.L.; Liu, Q.S.; Kang, H.X.; Liu, X.M.; Chen, X.P.; Peng, Y.F.; Li, Y.H. Herbivore-induced rice resistance against rice blast mediated by salicylic acid. Insect Sci. 2020, 27, 49–57. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, X.; Tzin, V.; Peng, Y.; Romeis, J.; Li, Y. Plant breeding involving genetic engineering does not result in unacceptable unintended effects in rice relative to conventional cross-breeding. Plant J. 2020, 103, 2236–2249. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Volodarsky, D.; Leviatan, N.; Otcheretianski, A.; Fluhr, R. HORMONOMETER: A tool for discerning transcript signatures of hormone action in the Arabidopsis transcriptome. Plant Physiol. 2009, 150, 1796–1805. [Google Scholar] [CrossRef] [PubMed]
- Šimura, J.; Antoniadi, I.; Široká, J.; Tarkowská, D.; Strnad, M.; Ljung, K.; Novák, O. Plant hormonomics: Multiple phytohormone profiling by targeted metabolomics. Plant Physiol. 2018, 177, 476–489. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Tzin, V.; Hojo, Y.; Strickler, S.R.; Bartsch, L.J.; Archer, C.M.; Ahern, K.R.; Zhou, S.; Christensen, S.A.; Galis, I.; Mueller, L.A.; et al. Rapid defense responses in maize leaves induced by Spodoptera exigua caterpillar feeding. J. Exp. Bot. 2017, 68, 4709–4723. [Google Scholar] [CrossRef]
- Guo, J.; Qi, J.; He, K.; Wu, J.; Bai, S.; Zhang, T.; Zhao, J.; Wang, Z. The Asian corn borer Ostrinia furnacalis feeding increases the direct and indirect defence of mid-whorl stage commercial maize in the field. Plant Biotechnol. J. 2019, 17, 88–102. [Google Scholar] [CrossRef]
- Li, J.; Zhu, L.; Hull, J.J.; Liang, S.; Daniell, H.; Jin, S.; Zhang, X. Transcriptome analysis reveals a comprehensive insect resistance response mechanism in cotton to infestation by the phloem feeding insect Bemisia tabaci (whitefly). Plant Biotechnol. J. 2016, 14, 1956–1975. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Sun, G.; Wang, L.; Zhao, C.; Hettenhausen, C.; Schuman, M.C.; Baldwin, I.T.; Li, J.; Song, J.; Liu, Z.; et al. Oral secretions from Mythimna separata insects specifically induce defence responses in maize as revealed by high-dimensional biological data. Plant Cell Environ. 2016, 39, 1749–1766. [Google Scholar] [CrossRef] [PubMed]
- Tzin, V.; Fernandez-Pozo, N.; Richter, A.; Schmelz, E.A.; Schoettner, M.; Schafer, M.; Ahern, K.R.; Meihls, L.N.; Kaur, H.; Huffaker, A.; et al. Dynamic maize responses to aphid feeding are revealed by a time series of transcriptomic and metabolomic assays. Plant Physiol. 2015, 169, 1727–1743. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.R.; Avramova, M.; Canham, J.; Higgins, P.; Bilkey, N.; Mugford, S.T.; Pitino, M.; Toyota, M.; Gilroy, S.; Miller, A.J.; et al. Interplay of plasma membrane and vacuolar ion channels, together with BAK1, elicits rapid cytosolic calcium elevations in arabidopsis during aphid feeding. Plant Cell 2017, 29, 1460–1479. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Fan, M.; Yang, M.; Zhao, J.; Zhang, W.; Su, Y.; Xiao, L.; Deng, H.; Xie, D. Injury activates Ca2+/calmodulin-dependent phosphorylation of JAV1-JAZ8-WRKY51 complex for jasmonate biosynthesis. Mol. Cell 2018, 70, 136–149.e137. [Google Scholar] [CrossRef] [PubMed]
- Vadassery, J.; Scholz, S.S.; Mithofer, A. Multiple calmodulin-like proteins in Arabidopsis are induced by insect-derived (Spodoptera littoralis) oral secretion. Plant Signal Behav. 2012, 7, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Scholz, S.S.; Vadassery, J.; Heyer, M.; Reichelt, M.; Bender, K.W.; Snedden, W.A.; Boland, W.; Mithofer, A. Mutation of the Arabidopsis calmodulin-like protein CML37 deregulates the jasmonate pathway and enhances susceptibility to herbivory. Mol. Plant 2014, 7, 1712–1726. [Google Scholar] [CrossRef]
- Vadassery, J.; Reichelt, M.; Hause, B.; Gershenzon, J.; Boland, W.; Mithofer, A. CML42-mediated calcium signaling coordinates responses to Spodoptera herbivory and abiotic stresses in Arabidopsis. Plant Physiol. 2012, 159, 1159–1175. [Google Scholar] [CrossRef]
- Heyer, M.; Scholz, S.S.; Voigt, D.; Reichelt, M.; Aldon, D.; Oelmuller, R.; Boland, W.; Mithofer, A. Herbivory-responsive calmodulin-like protein CML9 does not guide jasmonate-mediated defenses in Arabidopsis thaliana. PLoS ONE 2018, 13, e0197633. [Google Scholar] [CrossRef]
- Qi, J.; Malook, S.U.; Shen, G.; Gao, L.; Zhang, C.; Li, J.; Zhang, J.; Wang, L.; Wu, J. Current understanding of maize and rice defense against insect herbivores. Plant Divers. 2018, 40, 189–195. [Google Scholar] [CrossRef]
- Ye, M.; Luo, S.M.; Xie, J.F.; Li, Y.F.; Xu, T.; Liu, Y.; Song, Y.Y.; Zhu-Salzman, K.; Zeng, R.S. silencing COI1 in rice increases susceptibility to chewing insects and impairs inducible defense. PLoS ONE 2012, 7, e36214. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Robert, C.A.; Riemann, M.; Cosme, M.; Mene-Saffrane, L.; Massana, J.; Stout, M.J.; Lou, Y.; Gershenzon, J.; Erb, M. Induced jasmonate signaling leads to contrasting effects on root damage and herbivore performance. Plant Physiol. 2015, 167, 1100–1116. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.G.; Du, M.H.; Turlings, T.C.; Cheng, J.A.; Shan, W.F. Exogenous application of jasmonic acid induces volatile emissions in rice and enhances parasitism of Nilaparvata lugens eggs by the parasitoid Anagrus nilaparvatae. J. Chem. Ecol. 2005, 31, 1985–2002. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Glauser, G.; Lou, Y.; Erb, M.; Hu, L. Molecular dissection of early defense signaling underlying volatile-mediated defense regulation and herbivore resistance in rice. Plant Cell 2019, 31, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, J.; Li, J.; Zhou, G.; Wang, Q.; Bian, W.; Erb, M.; Lou, Y. Prioritizing plant defence over growth through WRKY regulation facilitates infestation by non-target herbivores. eLife 2015, 4, e04805. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Ye, M.; Li, R.; Zhang, T.; Zhou, G.; Wang, Q.; Lu, J.; Lou, Y. The rice transcription factor WRKY53 suppresses herbivore-induced defenses by acting as a negative feedback modulator of mitogen-activated protein kinase activity. Plant Physiol. 2015, 169, 2907–2921. [Google Scholar] [CrossRef]
- Huangfu, J.; Li, J.; Li, R.; Ye, M.; Kuai, P.; Zhang, T.; Lou, Y. The Transcription factor OsWRKY45 negatively modulates the resistance of rice to the brown planthopper Nilaparvata lugens. Int. J. Mol. Sci. 2016, 17, 697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fu, Y.; Wang, Q.; Liu, X.; Li, Q.; Chen, J. Transcriptome analysis reveals rapid defence responses in wheat induced by phytotoxic aphid Schizaphis graminum feeding. BMC Genom. 2020, 21, 339. [Google Scholar] [CrossRef] [PubMed]
- Schwachtje, J.; Baldwin, I.T. Why does herbivore attack reconfigure primary metabolism? Plant Physiol. 2008, 146, 845–851. [Google Scholar] [CrossRef]
- Zhou, S.; Lou, Y.R.; Tzin, V.; Jander, G. Alteration of Plant Primary Metabolism in Response to Insect Herbivory. Plant Physiol. 2015, 169, 1488–1498. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, Q.; Du, L.; Hallerman, E.M.; Li, Y. Transcriptomic and Metabolomic Responses of Rice Plants to Cnaphalocrocis medinalis Caterpillar Infestation. Insects 2020, 11, 705. https://doi.org/10.3390/insects11100705
Wang Y, Liu Q, Du L, Hallerman EM, Li Y. Transcriptomic and Metabolomic Responses of Rice Plants to Cnaphalocrocis medinalis Caterpillar Infestation. Insects. 2020; 11(10):705. https://doi.org/10.3390/insects11100705
Chicago/Turabian StyleWang, Yuqi, Qingsong Liu, Lixiao Du, Eric M. Hallerman, and Yunhe Li. 2020. "Transcriptomic and Metabolomic Responses of Rice Plants to Cnaphalocrocis medinalis Caterpillar Infestation" Insects 11, no. 10: 705. https://doi.org/10.3390/insects11100705
APA StyleWang, Y., Liu, Q., Du, L., Hallerman, E. M., & Li, Y. (2020). Transcriptomic and Metabolomic Responses of Rice Plants to Cnaphalocrocis medinalis Caterpillar Infestation. Insects, 11(10), 705. https://doi.org/10.3390/insects11100705