High Prevalence of a Newly Discovered Wutai Mosquito Phasivirus in Mosquitoes from Rio de Janeiro, Brazil


 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
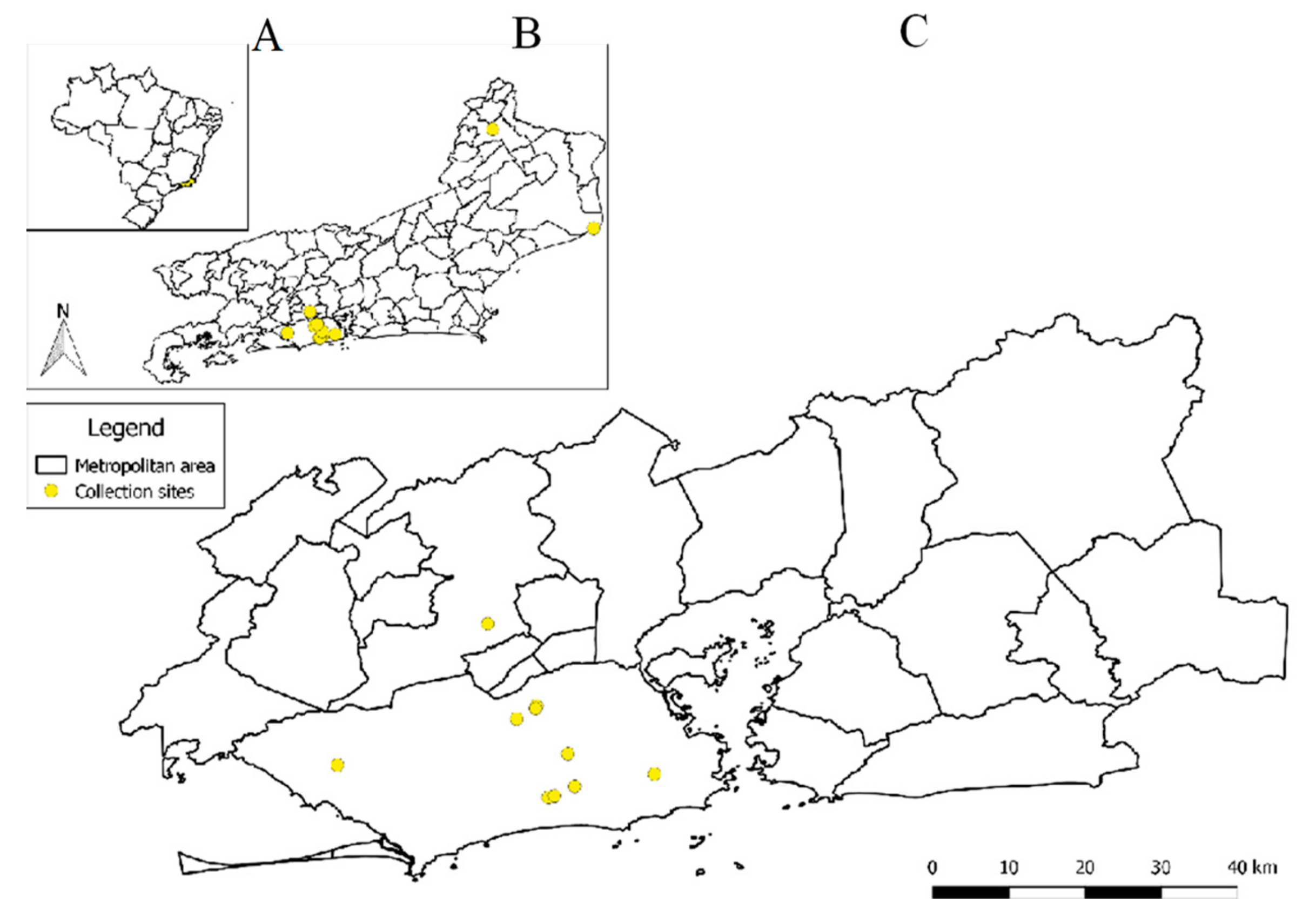
2.1. Sample Collection
2.2. Metagenomics Analysis
2.3. Wutai Mosquito Phasivirus RNA Detection by RT-PCR
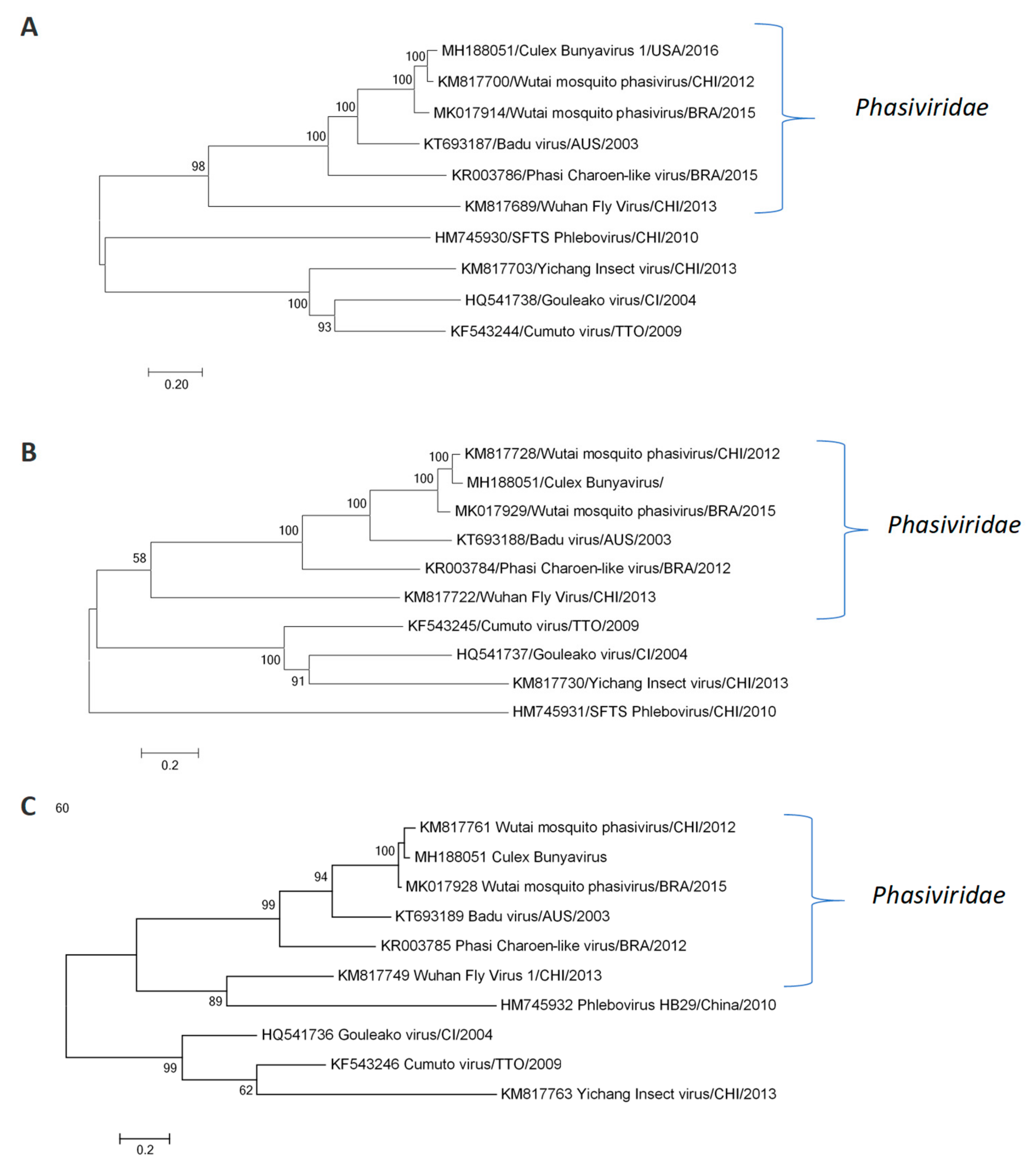
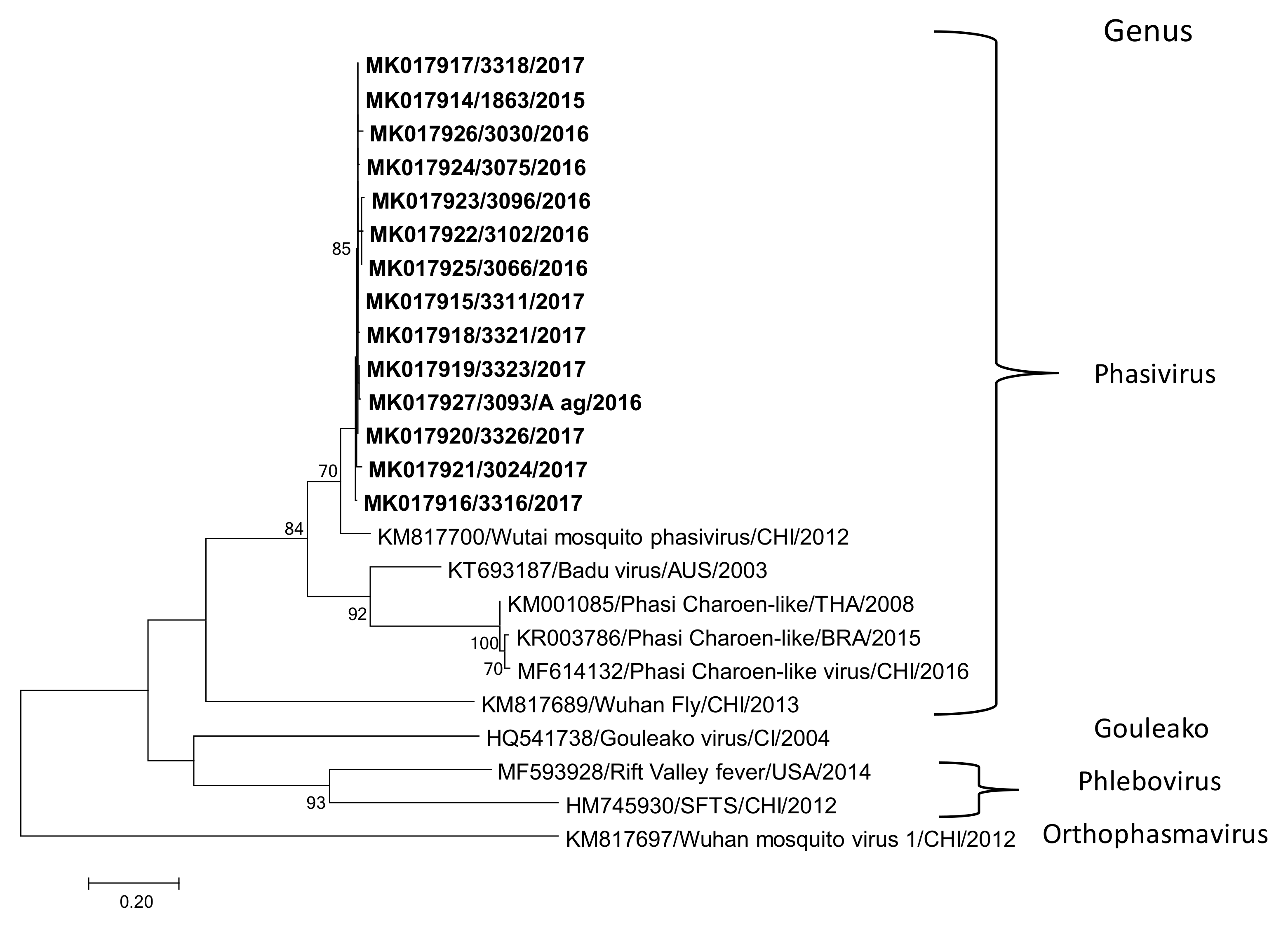
2.4. Nucleotide Sequence Determination and Phylogenetic Analysis
3. Results
3.1. Complete Genome of Wutai Mosquito Phasivirus Detected in Rio de Janeiro
3.2. Wutai Mosquito Phasisvirus Prevalence in Rio de Janeiro and Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Mazel-Sanchez, B.; Elliott, R.M. Evolution of the Bunyamwera Virus Polymerase to Accommodate Deletions within Genomic Untranslated Region Sequences. J. Virol. 2015, 89, 3957–3964. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Gowen, B.B.; Hickerson, B.T. Hemorrhagic fever of bunyavirus etiology: Disease models and progress towards new therapies. J. Microbiol. 2017, 55, 183–195. [Google Scholar] [CrossRef] [PubMed]
- De Souza, W.M.; Acrani, G.O.; Romeiro, M.F.; Reis, O.; Tolardo, A.L.; da Silva, S.P.; de Almeida Medeiros, D.B.; Varela, M.; Nunes, M.R.T.; Figueiredo, L.T.M. Molecular characterization of Capim and Enseada orthobunyaviruses. Infect. Genet. Evol. 2016, 40, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.F.; Serra, O.P.; da Silva Heinen, L.B.; Zuchi, N.; de Souza, V.C.; Naveca, F.G.; dos Santos, M.A.M.; Slhessarenko, R.D. Detection of Oropouche virus segment S in patients and inCulex quinquefasciatus in the state of Mato Grosso, Brazil. Mem. Inst. Oswaldo Cruz. 2015, 110, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Mourão, M.P.G.; de Souza Bastos, M.; de Figueiredo, R.M.P.; de Lima Gimaque, J.B.; do Carmo Rodrigues Alves, V.; das Graças Gomes Saraiva, M.; Figueiredo, M.L.G.; Ramasawmy, R.; Nogueira, M.L.; Figueiredo, L.T.M. Arboviral diseases in the western brazilian amazon: A perspective and analysis from a tertiary health & research center in manaus, state of Amazonas. Rev. Soc. Bras. Med. Trop. 2015, 48. [Google Scholar] [CrossRef]
- Nunes, M.R.T.; Travassos da Rosa, A.P.A.; Weaver, S.C.; Tesh, R.B.; Vasconcelos, P.F.C. Molecular Epidemiology of Group C Viruses (Bunyaviridae, Orthobunyavirus) Isolated in the Americas. J. Virol. 2005, 79, 10561–10570. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.B.; Luiz, A.P.M.F.; Fagundes, A.; Pinto, C.A.; Bonjardim, C.A.; Trindade, G.S.; Kroon, E.G.; Abrahão, J.S.; Ferreira, P.C. Evidence of apeu virus infection in wild monkeys, Brazilian Amazon. Am. J. Trop. Med. Hyg. 2016, 94, 494–496. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marklewitz, M.; Handrick, S.; Grasse, W.; Kurth, A.; Lukashev, A.; Drosten, C.; Ellerbrok, H.; Leendertz, F.H.; Pauli, G.; Junglen, S. Gouleako Virus Isolated from West African Mosquitoes Constitutes a Proposed Novel Genus in the Family Bunyaviridae. J. Virol. 2011, 85, 9227–9234. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Peters, J.; Warrilow, D.; McLean, B.J.; Watterson, D.; Colmant, A.M.G.; van den Hurk, A.F.; Hall-Mendelin, S.; Hastie, M.L.; Gorman, J.J.; Harrison, J.J.; et al. Discovery and characterisation of a new insect-specific bunyavirus from Culex mosquitoes captured in northern Australia. Virology 2016, 489, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, E.R.G.R.; Olmo, R.P.; Paro, S.; Ferreira, F.V.; De Faria, I.J.D.S.; Todjro, Y.M.H.; Lobo, F.P.; Kroon, E.G.; Meignin, C.; Gatherer, D.; et al. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res. 2015, 43, 6191–6206. [Google Scholar] [CrossRef] [PubMed]
- Principais mosquitos de importância sanitária no Brasil. Available online: http://books.scielo.org (accessed on 7 March 2018).
- Börstler, J.; Jöst, H.; Garms, R.; Krüger, A.; Tannich, E.; Becker, N.; Schmidt-Chanasit, J.; Lühken, R. Host-feeding patterns of mosquito species in Germany. Parasit. Vectors. 2016, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.A.; Bielefeldt-Ohmann, H.; McLean, B.J.; O’Brien, C.A.; Colmant, A.M.G.; Piyasena, T.B.H.; Harrison, J.J.; Newton, N.D.; Barnard, R.T.; Prow, N.A.; et al. Commensal viruses of mosquitoes: Host restriction, transmission, and interaction with arboviral pathogens. Evol. Bioinform. Online 2016, 12, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.D.; Ciota, A.T. Dissecting vectorial capacity for mosquito-borne viruses. Curr. Opin. Virol. 2015, 15, 112–118. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locations | Males n | Pools n (pos) | Non-Engorged Females n | Pools n (pos) | Engorged Females | |
|---|---|---|---|---|---|---|
| n | Pos n (%) | |||||
| 1 | 107 | 12 (6) | 74 | 11 (8) | 0 | - |
| 2 | 180 | 4 (2) | 77 | 4 (1) | 47 | 1 (2.1%) |
| 3 | 21 | 1 (1) | 55 | 4 (0) | 21 | 0 |
| 4 | 0 | - | 0 | - | 2 | 0 |
| 5 | 0 | - | 15 | 1 (0) | 21 | 0 |
| 6 | 49 | 4 (2) | 43 | 4 (3) | 0 | - |
| 7 | 5 | 1 (1) | 12 | 2 (1) | 6 | 0 |
| 8 | 0 | - | 0 | - | 3 | 0 |
| 9 | 32 | 3 (0) | 18 | 2 (1) | 31 | 0 |
| 10 | 347 | 2 (2) | 98 | 1 (1) | 15 | 0 |
| 11 | 0 | - | 0 | - | 74 | 8 (10.81%) |
| 12 | 83 | 4 (3) | 26 | 2(0) | 0 | - |
| 13 | 0 | - | 0 | - | 3 | 0 |
| Total | 824 | 31 (17) | 418 | 32 (15) | 223 | 9 (4.03%) |
| Locations | Males n | Pools n (pos) | Non-Engorged Females n | Pools n (pos) | Engorged Females | |
|---|---|---|---|---|---|---|
| n | Pos n (%) | |||||
| 2 | 64 | 13 (0) | 22 | 11 (0) | 0 | - |
| 3 | 33 | 10 (0) | 20 | 7 (0) | 0 | - |
| 4 | 0 | - | 5 | 3 (0) | 0 | - |
| 7 | 13 | 7 (0) | 16 | 7 (0) | 0 | - |
| 9 | 26 | 9 (0) | 28 | 11 (0) | 1 | 0 |
| 10 | 14 | 4 (1) | 157 | 4 (0) | 0 | - |
| 13 | 0 | 0 | 0 | 0 | 2 | 0 |
| Total | 150 | 43 (1) | 248 | 43 (0) | 3 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, M.S.; Ayllón, T.; Malirat, V.; Câmara, D.C.P.; Giordano Dias, C.M.; Louzada, G.; Fernandes-Ferreira, D.; Medronho, R.d.A.; Campos Acevedo, R. High Prevalence of a Newly Discovered Wutai Mosquito Phasivirus in Mosquitoes from Rio de Janeiro, Brazil. Insects 2019, 10, 135. https://doi.org/10.3390/insects10050135
Ribeiro MS, Ayllón T, Malirat V, Câmara DCP, Giordano Dias CM, Louzada G, Fernandes-Ferreira D, Medronho RdA, Campos Acevedo R. High Prevalence of a Newly Discovered Wutai Mosquito Phasivirus in Mosquitoes from Rio de Janeiro, Brazil. Insects. 2019; 10(5):135. https://doi.org/10.3390/insects10050135
Chicago/Turabian StyleRibeiro, Mário Sérgio, Tania Ayllón, Viviana Malirat, Daniel Cardoso Portela Câmara, Cristina Maria Giordano Dias, Guilherme Louzada, Davis Fernandes-Ferreira, Roberto de Andrade Medronho, and Renata Campos Acevedo. 2019. "High Prevalence of a Newly Discovered Wutai Mosquito Phasivirus in Mosquitoes from Rio de Janeiro, Brazil" Insects 10, no. 5: 135. https://doi.org/10.3390/insects10050135
APA StyleRibeiro, M. S., Ayllón, T., Malirat, V., Câmara, D. C. P., Giordano Dias, C. M., Louzada, G., Fernandes-Ferreira, D., Medronho, R. d. A., & Campos Acevedo, R. (2019). High Prevalence of a Newly Discovered Wutai Mosquito Phasivirus in Mosquitoes from Rio de Janeiro, Brazil. Insects, 10(5), 135. https://doi.org/10.3390/insects10050135