Effects of Different Pretreatments of DNA Extraction from Dried Specimens of Ladybird Beetles (Coleoptera: Coccinellidae)


Abstract
1. Introduction
2. Materials and Methods
2.1. Specimens
2.2. Pretreatments
2.3. DNA Extraction and PCR Amplification
2.4. Sequences Composition and Phylogenetic Analysis
3. Results
3.1. Measurement of DNA Purity and Concentration
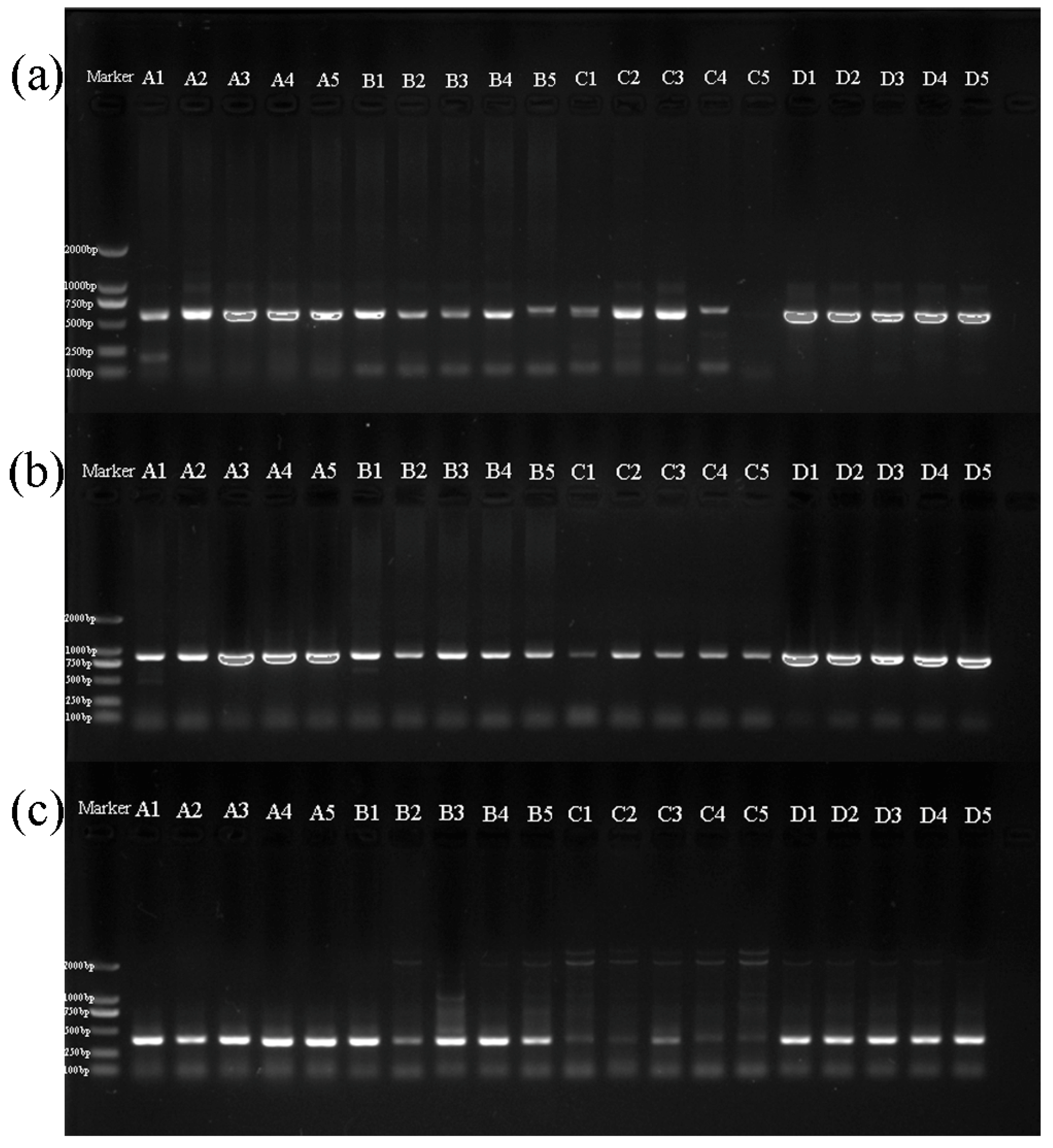
3.2. Amplification of Sequences
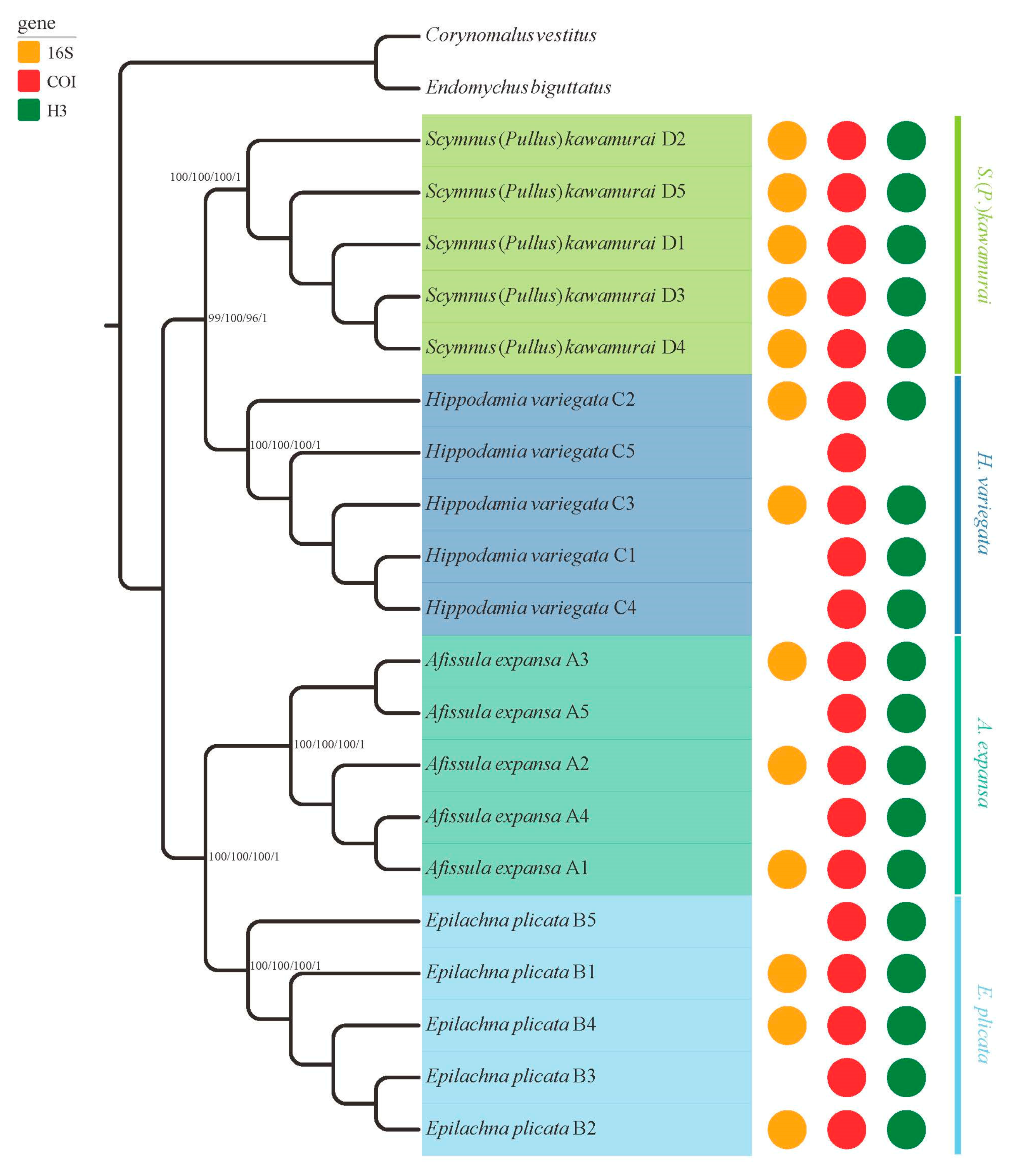
3.3. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Houdt, J.K.J.; Breman, F.C.; Virgilio, M.; De Meyer, M. Recovering full DNA barcodes from natural history collections of Tephritid fruitflies (Tephritidae, Diptera) using mini barcodes. Mol. Ecol. Resour. 2010, 10, 459–465. [Google Scholar] [CrossRef]
- Wandeler, P.; Hoeck, P.E.; Keller, L.F. Back to the future: Museum specimens in population genetics. Trends Ecol. Evol. 2007, 22, 634–642. [Google Scholar] [CrossRef]
- Parham, J.F.; Stuart, B.L.; Bour, R.; Fritz, U. Evolutionary distinctiveness of the extinct Yunnan box turtle (Cuora yunnanensis) revealed by DNA from an old museum specimen. Proc. R. Soc. B Biol. Sci. 2004, 271, S391–S394. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Siedel, H.; Hofreiter, M. Nondestructive DNA extraction method for mitochondrial DNA analyses of museum specimens. Biotechniques 2004, 36, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Horváth, M.B.; Martínez-Cruz, B.; Negro, J.J.; Kalmár, L.; Godoy, J.A. An overlooked DNA source for non-invasive genetic analysis in birds. J. Avian Biol. 2005, 36, 84–88. [Google Scholar] [CrossRef]
- Harper, G.L.; Maclean, N.; Goulson, D. Analysis of museum specimens suggests extreme genetic drift in the adonis blue butterfly (Polyommatus bellargus). Biol. J. Linn. Soc. 2006, 88, 447–452. [Google Scholar] [CrossRef]
- An, W.T.; Ren, G.D.; Liu, F.S. Genomic DNA extracting and systematic study of the dry preserved tribe Platyopini specimen of China. J. Hebei Univ. 2010, 30, 190–195. (In Chinese) [Google Scholar]
- Li, J.L.; Zheng, S.Z.; Cai, P.; Zhan, G.H.; Gao, Y. Effects of different pretreatment on DNA extraction from dried specimen of insects. Genom. Appl. Biol. 2015, 34, 396–402. (In Chinese) [Google Scholar]
- Pu, M.H.; Chen, X.X.; He, J.H. Extraction of genome DNA from dried specimens of Hymenopteran insects. Zool. Syst. 2002, 27, 672–676. (In Chinese) [Google Scholar]
- Zhang, D.H.; Zhou, K.Y.; Sun, H.Y. Comparison of analytical methods for extracting genomic DNA form ethanol-preserved animal specimens. J. Biol. 2004, 21, 46–48. (In Chinese) [Google Scholar]
- Zimmermann, J.; Hajibabaei, M.; Blackburn, D.C.; Hanken, J.; Cantin, E.; Posfai, J.; Evans, T.C. DNA damage in preserved specimens and tissue samples: A molecular assessment. Front. Zool. 2008, 5, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Dick, M.; Bridge, D.M.; Wheeler, W.C.; Desalle, R. Collection and storage of invertebrate samples. Methods Enzymol. 1993, 224, 51–65. [Google Scholar]
- Yang, Z.H.; Rannala, B. Molecular phylogenetics: Principles and practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef]
- Magro, A.; Lecompte, E.; Magne, F.; Hemptinne, J.L.; Crouau-Roy, B. Phylogeny of ladybirds (Coleoptera: Coccinellidae): Are the subfamilies monophyletic? Mol. Phylogenet. Evol. 2010, 54, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, M.J.; Dodsworth, S.; Culverwell, C.L.; Bocak, L.; Ahrens, D.; Littlewood, D.T.; Vogler, A.P. Why barcode? High-throughput multiplex sequencing of mitochondrial genomes for molecular systematics. Nucleic Acids Res. 2010, 38, e197–e210. [Google Scholar] [CrossRef]
- Robertson, J.A.; Ślipiński, A.; Hiatt, K.; Miller, K.B.; Whiting, M.F.; McHugh, J.V. Molecules, morphology and minute hooded beetles: A phylogenetic study with implications for the evolution and classification of Corylophidae (Coleoptera: Cucujoidea). Syst. Entomol. 2013, 38, 209–232. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Robertson, J.A.; Ślipiński, A.; Moulton, M.; Shockley, F.W.; Giorgi, A.; Lord, N.P.; McKenna, D.D.; Tomaszewska, W.; Forrester, J.; Miller, K.B.; et al. Phylogeny and classification of Cucujoidea and the recognition of a new superfamily Coccinelloidea (Coleoptera: Cucujiformia). Syst. Entomol. 2015, 40, 745–778. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Che, L.H.; Li, Y.; Pang, H.; Ślipiński, A.; Zhang, P. Evolutionary history of Coleoptera revealed by extensive sampling of genes and species. Nat. Commun. 2018, 9, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*, Phylogenetic Analysis Using Parsimony (and Other Methods); Version 4.0; Sinauer Associates, Inc.: Sunderland, MA, USA, 2002. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. Partitionfinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Tracer v1.6. 2013. Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 18 November 2018).
- Pääbo, S.; Poinar, H.; Serre, D.; Jaenicke-Després, V.; Hebler, J.; Rohland, N.; Hofreiter, M. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004, 38, 645–679. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A. DNA from museum specimens. In Ancient DNA: Recovery and Analysis of Genetic Material from Paleontological, Archeological, Museum, Medical and Forensic Specimens; Herrmann, B., Herrmann, S., Eds.; Springer: New York, NY, USA, 1994. [Google Scholar]
- Sefc, K.M.; Payne, R.B.; Sorenson, M.D. Single base errors in PCR products from avian museum specimens and their effect on estimates of historical genetic diversity. Conserv. Genet. 2007, 8, 879–884. [Google Scholar] [CrossRef]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; Haeseler, A.V.; Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Szawaryn, K.; Bocak, L.; Ślipiński, A.; Escalona, H.E.; Tomaszewska, W. Phylogeny and evolution of phytophagous ladybird beetles (Coleoptera: Coccinellidae: Epilachnini), with recognition of new genera. Syst. Entomol. 2015, 40, 547–569. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Pretreatment | Code | Collection Date | Species | GenBank Accession | ||
|---|---|---|---|---|---|---|
| 16S | COI | H3 | ||||
| 0.9% NaCl buffer | A1 | 2009 | Afissula expansa | MK138697 | MK190427 | MK190447 |
| PBS buffer | A2 | 2009 | A. expansa | MK138698 | MK190428 | MK190448 |
| STE buffer | A3 | 2009 | A. expansa | MK138699 | MK190429 | MK190449 |
| Sterile water | A4 | 2009 | A. expansa | - | MK190430 | MK190450 |
| CK | A5 | 2009 | A. expansa | - | MK190431 | MK190451 |
| 0.9% NaCl buffer | B1 | 2009 | Epilachna plicata | MK138700 | MK190432 | MK190452 |
| PBS buffer | B2 | 2009 | E. plicata | MK138701 | MK190433 | MK190453 |
| STE buffer | B3 | 2009 | E. plicata | - | MK190434 | MK190454 |
| Sterile water | B4 | 2009 | E. plicata | MK138702 | MK190435 | MK190455 |
| CK | B5 | 2009 | E. plicata | - | MK190436 | MK190456 |
| 0.9% NaCl buffer | C1 | 2008 | Hippodamia variegata | - | MK190437 | MK190457 |
| PBS buffer | C2 | 2008 | H. variegata | MK138703 | MK190438 | MK190458 |
| STE buffer | C3 | 008 | H. variegata | MK138704 | MK190439 | MK190459 |
| Sterile water | C4 | 2008 | H. variegata | - | MK190440 | MK190460 |
| CK | C5 | 2008 | H. variegata | - | MK190441 | - |
| 0.9% NaCl buffer | D1 | 2012 | Scymnus (Pullus) kawamurai | MK138705 | MK190442 | MK190461 |
| PBS buffer | D2 | 2012 | S. (P.) kawamurai | MK138706 | MK190443 | MK190462 |
| STE buffer | D3 | 2012 | S. (P.) kawamurai | MK138707 | MK190444 | MK190463 |
| Sterile water | D4 | 2012 | S. (P.) kawamurai | MK138708 | MK190445 | MK190464 |
| CK | D5 | 2012 | S. (P.) kawamurai | MK138709 | MK190446 | MK190465 |
| Marker | Primer Name | Primer Sequence (5′-3′) | Reference |
|---|---|---|---|
| COI | Jerry | CAACATTTATTTTGATTTTTT | [15] |
| Spat | GCACTAWTCTGCCATATTAGA | [15] | |
| 16S | 16S A | CGCCTGTTTATCAAAAACAT | [16] |
| 16S B | CTCCGGTTTGAACTCAGATCA | [16] | |
| H3 | H3F-1 | CAGAAAGTCGACCGGAGGCAAG | This study |
| H3R-1 | GCGTTTCGCGTGAATGGCG | This study |
| Pretreatment | Code | OD260/280 | Concentration (ng/µL) |
|---|---|---|---|
| 0.9% NaCl buffer | A1 | 1.936 | 433.159 |
| B1 | 1.798 | 44.513 | |
| C1 | 2.075 | 61.52 | |
| D1 | 1.899 | 69.99 | |
| PBS buffer | A2 | 1.966 | 539.647 |
| B2 | 1.929 | 202.086 | |
| C2 | 2.067 | 34.331 | |
| D2 | 1.924 | 20.108 | |
| STE buffer | A3 | 1.942 | 464.019 |
| B3 | 1.823 | 93.246 | |
| C3 | 2.074 | 31.527 | |
| D3 | 1.864 | 26.774 | |
| Sterile water | A4 | 2.003 | 678.726 |
| B4 | 1.98 | 52.972 | |
| C4 | 2.071 | 32.554 | |
| D4 | 1.947 | 17.117 | |
| CK | A5 | 1.983 | 537.078 |
| B5 | 2.007 | 26.408 | |
| C5 | 2.098 | 14.513 | |
| D5 | 2.090 | 23.265 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.; Xie, X.; Liang, X.; Wang, X.; Chen, X. Effects of Different Pretreatments of DNA Extraction from Dried Specimens of Ladybird Beetles (Coleoptera: Coccinellidae). Insects 2019, 10, 91. https://doi.org/10.3390/insects10040091
Huang W, Xie X, Liang X, Wang X, Chen X. Effects of Different Pretreatments of DNA Extraction from Dried Specimens of Ladybird Beetles (Coleoptera: Coccinellidae). Insects. 2019; 10(4):91. https://doi.org/10.3390/insects10040091
Chicago/Turabian StyleHuang, Weidong, Xiufeng Xie, Xinyue Liang, Xingmin Wang, and Xiaosheng Chen. 2019. "Effects of Different Pretreatments of DNA Extraction from Dried Specimens of Ladybird Beetles (Coleoptera: Coccinellidae)" Insects 10, no. 4: 91. https://doi.org/10.3390/insects10040091
APA StyleHuang, W., Xie, X., Liang, X., Wang, X., & Chen, X. (2019). Effects of Different Pretreatments of DNA Extraction from Dried Specimens of Ladybird Beetles (Coleoptera: Coccinellidae). Insects, 10(4), 91. https://doi.org/10.3390/insects10040091