Abstract
The complex composition of lubricating base oils makes it difficult to analyze the influence of specific molecular structure on lubricating performance. To achieve this target, nine kinds of poly α-olefin molecules with different structure characteristics were designed, which prepared the lubricant models. Molecular dynamic simulation was used to analyze the tribological performance under pressure of 500 MPa, temperature 353 K, and shear velocity of 20 m/s; the volume compression and shear stress of lubricant films were obtained. Molecular volume, adsorption energies, radius of gyration, and mean square displacement were used to analyze the relationship between molecular structure and lubricant performance. Results show that the characteristic of the Iso and Mid type have the best friction reduction performance. The molecules of the Iso structure have the highest oil film thickness and the best load-bearing performance. The radius of gyration increases with the shear simulation for most of the molecules. The adsorption energy of End is the highest, and the Mid is the smallest. Among the nine molecules, C20Iso shows excellent performance both in load-bearing and friction reduction, which provides a reference for the molecular design of high-performance lubricant base oils.
1. Introduction
Reducing friction loss is one of the key measures to reduce fuel consumption in internal combustion engines, which improves fuel efficiency. High-performance lubricant is a crucial factor in reducing friction and minimizing wear [1]. As the performance of internal combustion engines has improved rapidly in recent years, the burst pressure and rotational speed have increased, putting higher demands on lubricants.
Lubricants are composed of base oils and various additives. Base oils are usually processed and refined from crude oil. Still, they can also be produced through synthetic processes, and their properties can be improved by additives, which interact with the friction surface and base oil to meet various needs through chemical or physical effects. The properties of the base oil have an important influence on the lubricant’s performance. These properties include viscosity, viscosity index, flash point, pour point, volatility, oxidative stability, thermal stability, etc. The molecular structure and composition of base oils directly affect these properties. Therefore, it is essential to study the effect of the base oil molecular structure on lubricating properties to improve the performance of lubricants.
Sarpal [2] proposed that the quantitative distribution and type of different classes of hydrocarbons determine the physicochemical behavior of a lubricant system and that specific methyl branches along the alkyl carbon chain can be produced by catalytic isomerization of feedstocks to produce base oils with desired properties. Zhang [3] compared the tribological properties of base oils such as synthetic esters and mineral oils using viscosity, friction, and wear, which shows that friction and lubricant molecular structure may be related. Increasing the number of branched chains or carbon atoms for esters with branched structures can improve the low-friction performance of ester lubricants. Airey [4] used a micro tractor to plot Stribeck curves under representative oil system conditions by varying test temperature. The results show that molecular structure significantly affects measured friction in all lubrication states, with friction decreasing as chain length increases and the number of ester groups increases.
In addition to the experimental analysis of the tribological and physicochemical properties of lubricants, molecular dynamics simulation is also an essential method for analyzing the molecular conformational relationships of lubricant base oils. The molecular dynamics method can simulate organic lubricant molecules and nanoparticles, exploring the tribological properties in combination with friction surfaces of different morphologies [5,6]. Washizu [7] conducted molecular dynamics studies of the tribological properties of hydrocarbon fluids under elastohydrodynamic lubrication conditions, examining the effects of shear rate and lubricant film thickness, bringing fluid molecules related to momentum transfer to the forefront. Stephan [8] used classical molecular dynamics simulations to study the lubrication contact process to determine the entire range of the Stribeck curve, and methane and decane were used as lubricants in the study. The results of the simulations revealed that the formation of the friction film has an important influence on the contact process and that, in hydrodynamic lubrication, the tribological performance of the decane is better. In boundary lubrication, the friction coefficient is more significant than methane due to the different mechanisms of friction film formation. The mechanism is different; the decane’s friction coefficient is more extensive than that of methane. Cheng [9] used molecular dynamics simulation to investigate the load-bearing and lubrication properties of konjac glucomannan (KGM) solutions and revealed the lubrication mechanism of KGMs. The simulation results showed that when the number of KGMs is too large, the concentration of KGM molecules at the middle position of the liquid phase increases, and the flow resistance of the liquid phase layer increases, which leads to more considerable interfacial shear stresses, and the interfacial shear stresses increase with the increase in the shear rate.
Xue prepared the low-viscosity poly α-olefin (PAO) using α-olefins with different chain lengths. The relationship between molecular structure parameters and viscosity–temperature properties was studied by relative molecular weight (Mn), branch carbon number (L), and number of branches (n). Results indicate that the increased Mn improved the viscosity index, and a greater L results in a weaker growth trend [10]. In the experimental study above, as the PAO is a mixture of polymers with different polymerization degrees, the results lack the detailed analysis of each specific molecular structure.
Molecular dynamics simulation is an efficient way to study the friction reduction mechanism at the molecular level. Du conducted molecular dynamic (MD) simulations of lubricants composed of three alkanes, using a Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies (COMPASS) force field for calculations, and discussed the density distribution and final film thickness of lubricant films in non-operating and operating conditions. The results show that the viscosity shows an oscillating decreasing tendency with increasing temperature, the film thickness has almost the same rate of change with the pressure loading, and the peak density increases when the pressure increases [11]. Several previous studies have used bulk MD simulations to investigate the viscosity and visco-temperature relationship of PAO isomers [12]. It appears that PAO molecules with long, widely spaced branches should yield a higher viscosity number than those with short, closely spaced branches. Shi [13] used molecular dynamics simulation to study the synergistic effect of friction modifiers compounded with glycerol monolaurate (GML) and tri-n-octylamine (N888) in base oils. The adsorption and shear behaviors showed that the three-chain N888 molecules were dispersed between the long-chain GLM molecules to form adsorbent membranes coordinated with each other. The best lubrication effect and the lowest friction coefficient were achieved when the quantity ratio of GML to N888 was 3:1. Zheng [14] established non-equilibrium molecular dynamics (NEMD) simulations in a boundary friction system with mixed C4 alkanes and nanoparticles as a lubricant and discussed the effects of nanoparticles and fluid additions on surface contact and friction. It was found that nanoparticles acted as a ball-bearing-like effect between the contacting surfaces, which changed the sliding friction mode into a rolling friction mode, and this study contributes to the understanding of the friction behavior of a simple lubricant with or without nanoparticle additions in small spaces. Friction behavior of simple lubricants with or without nanoparticle addition. Pan [15] conducted MD simulations to explore the lubrication mechanism and boundary slip of squalane in the nanogap for two different film thicknesses and pressure ranges at 293 K. The molecular distribution, density, etc., of squalane are discussed, and the study results show that the lubricant atoms tend to form a laminar structure parallel to the wall. Still, the lubricant molecules are randomly oriented in the film in parallel-to-wall and perpendicular-to-wall orientations. Most squalane molecules show twisting and folding and extend to several atomic layers, so there is no slip between lubricant layers. Zheng [16] discussed the effects of lubricant molecular chain length and average load on friction and rough-peak contact through molecular dynamics simulations of constrained n-alkanes, which showed that long-chained n-alkanes provide more atoms in the monomolecular layer at the rough-peak contact interface than short-chained n-alkanes, resulting in a significant reduction in friction during rough-peak contact. Katsukawa [17] investigated the lubricating properties of the molecular lubricants PEE 15 and PEE 30 and the effects of temperature and carbon chain length on their lubricating properties in contact with iron and found that the friction coefficient is reduced when the temperature is higher or the carbon chain is shorter. The friction coefficient decreases as the temperature increases or the length of the carbon chain (number of branches) decreases. A study of Mehrnia shows that as the number of branches of the PAO molecule increases, the shear stresses and the wall slip increase. However, for a mixture lubricant containing different branched molecules, the wall slip decreases in comparison to a liquid consisting of only one kind of branched molecule [18]. Hou [19] investigated the effect of branched hydrocarbon molecules on the boundary lubrication of ZnO nanofluids using MD simulations and discussed the film structure and friction of the lubrication film at the atomic scale. The results showed that the higher number of PAO branched chains and the higher number of atoms can significantly reduce the friction in the case of non-uniform contact, with a maximum reduction of up to about 95%. With the increase in the number of PAO branched chains, the slip distance of the system increases with the increase in the number of PAO branched chains. Xu [20] performed reactive CGMD simulations under the ReaxFF force field to investigate the friction chemical interactions between PTFE and Fe. The results showed that the shear process leads to the breaking of the interfacial anchoring bonds as well as the chain disentanglement in the matrix, which in turn leads to the ordered reorientation of the molecular chains in the direction of sliding, thus reducing the friction. Zhang [21] investigated the effect of pressure and shear rate on friction and friction force coefficients. In contrast, the molecular dynamics method simulated van der Waals energy, shear stress, and solid film thickness changes in the lubrication region. It was concluded that the additives increased the van der Waals energy in the lubrication region and the thickness of the solid film while decreasing the shear stress, friction coefficient, and friction force of the Si3N4-GCr15 friction pair. The effect of graphene on the friction and lubrication properties of Si3N4-GCr15 is more significant with the increase in pressure and shear rate. Savio [22] used molecular dynamics to study the transition from ultra-thin lubrication to dry friction under high-pressure and high-shear conditions, where the molecular structure affects the ordering of the fluid and the resulting shear stresses, and localized film rupture is observed when rough peaks come into contact, with solid wall interactions leading to maximum shear stress. Xiong [23] used the molecular dynamics method to simulate the shear process of a lubricant system with a mixture of four molecules in the Fe-Al system and calculated the friction coefficient of the lubricant. The shear stress affects the adsorption morphology of the molecules, and the long-chain molecules are curled to some extent. With the increase in pressure perpendicular to the shear surface, the aggregation of lubricant molecules on the metal wall decreases, and the distribution of lubricant molecules between the metal walls is gradually homogenized.
Although various studies were conducted to study the structural-lubrication relationship of base oil molecules, the complex composition of lubricating base oils makes it difficult to analyze the influence of specific molecular structures on lubricating performance. Therefore, in this paper, molecular dynamics simulation is used to study the tribological performance of pure lubricating base oils composed of a single kind of molecule, and the structural characteristics of high-performance base oil molecules with low shear stress and high load-bearing performance were studied.
2. Materials and Methods
2.1. Definition of Molecular Structure
Lubricants are complex mixtures of alkane molecules of various structures. Cycloalkanes, aromatic hydrocarbons, and unsaturated hydrocarbons in lubricating oils will adversely affect the antioxidant and viscosity-temperature properties, so the lubricating base oils with good performance are composed of saturated alkanes. Poly α olefin (PAO) is ideal as a base oil due to its high viscosity index and thermally stable properties. Therefore, in this study, nine base oil molecules were designed based on PAO molecules. Alkanes containing 20–40 C atoms were used as the object of study, and the main difference in these molecules is the position of the carbon branches. Where the alkane molecule appears as a typical PAO molecule and presents a comb-like structure, the suffix is Iso. Where the molecule is branched in the middle of the main chain, the suffix is Mid. Where the molecule is branched in both segments of the main chain, the suffix is End. The lubrication model parameters are shown in Table 1.

Table 1.
Lubrication model parameters.
2.2. Lubrication Modeling
The different structurally characterized molecules mentioned above are established, and the Iso, Mid, and End structures are shown in Figure 1. It is noticeable that the molecular structure in Figure 1 is the one before geometry optimization, which can significantly reflect the differences in each molecule. After the molecular structure was optimized, due to the rotation and bending of the carbon chains, the differences were not as clear as shown in Figure 1. The molecules in Figure 1 were used to build the lubricant film after geometry optimization. Each base oil lubricant film was built with a single kind of molecule, and the lubricant film dimension is 60.2 × 40.1 × 40 Å3. To avoid the failure of modeling and incorrect spatial distribution of carbon chains, the original density is slightly lower, which is 0.6 g/cm3.
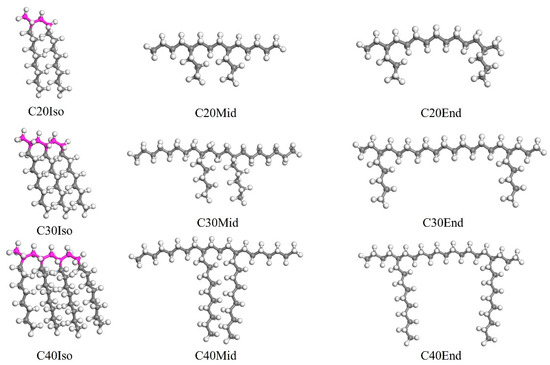
Figure 1.
The molecular structure of Iso, Mid, and End molecules before geometry optimization. The color of grey stand for carbon, the white is hydrogen, and the pink is the main chain of PAO.
The structure of lubricant layer is shown in Figure 2. Due to the different structural sizes of each characteristic molecule, if the same number of molecules is added to the lubricant film, the thickness of the lubricant film will be significantly different. To ensure that the lubrication film thicknesses of the molecular dynamics models are the same and reduce the influence of film thickness differences on the simulation results, the number of molecules in the C20, C30, and C40 lubrication models is gradually reduced. Subsequently, the model of lubricating lubricant film was combined with an iron layer with dimensions of 60.2 × 40.1 × 14.3 Å3 to form an iron-oil-iron model.
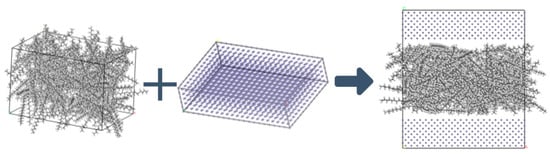
Figure 2.
Lubricant layer structure.
2.3. Computational Analysis
In this study, Materials Studio is used for these simulations. The COMPASS force field [24] is used for the whole system, the summation method of electrostatic and van der Waals is all-atom-based, and the simulation quality is ultra-fine with a time step of 1 fs. Once the model is built, the Geometry Optimization task is carried out in the Forcite module to optimize the molecular structure of the lubricant. The algorithm is smart, and the max iterations are 2000. Then the annealing procedure is carried out with the NVT ensemble, which represents a system in which the number of particles (N), volume (V), and temperature (T) remain constant. The Nose-Hoover thermostat [25,26] is used to control temperature. The annealing cycles are 20, with an initial temperature of 300 K and a mid-cycle temperature of 500 K. After the annealing procedure, the load is exerted on the lubricating system with Perl Script. The pressure of 500 MPa is set in the upper wall, and a counter force is exerted on the rest of the system. The compression is equilibrated with 0.5 ns, and an overall temperature of 353 K is set for the system, which is a representative temperature at lubrication conditions [27]. Finally, the Confined Shear task is carried out, the NVT ensemble is applied to the whole system [28,29], and the upper and lower iron atoms are moving in opposite directions with a relative speed of 20 m/s to simulate the friction of the lubricant layer. A total simulation of 6 ns is conducted; the first 1 ns was used to achieve the steady shear state, and the tribological results were obtained in the following 2–6 ns, and the confined shear diagram is shown in Figure 3. After simulation, the average shear stress, lubricant film compression distance, adsorption energies, radius of gyration, relative concentration, velocity profiles, and mean square displacement were analyzed.
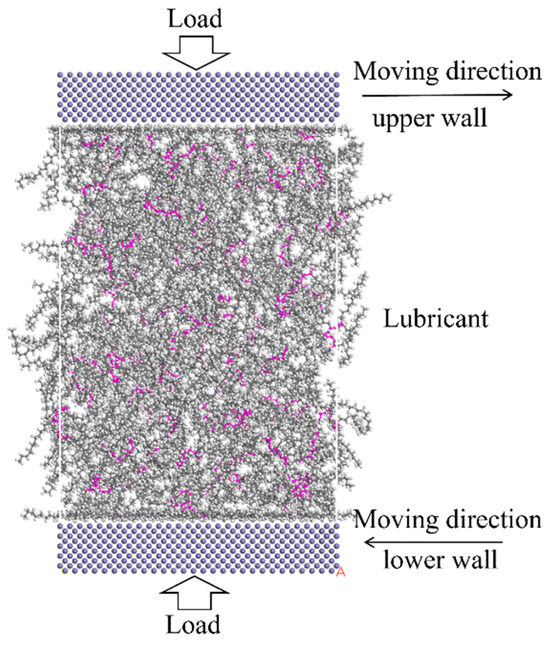
Figure 3.
Confined shear diagram of the lubricant system. The color of blue stand for iron, the grey stand for carbon, the white is hydrogen, and the pink is the main chain of PAO.
3. Results
3.1. Shear Stress
As shown in Figure 4, the smaller the molecule, the lower the shear stress. Generally, the Iso and Mid structures have smaller shear stress than the End structure. The average shear stress results from the simulation are C20End > C20Mid > C20Iso, C30End > C30Mid ≈ C30Iso, C40End > C40Iso ≈ C40Mid, and the shear stress of End structure molecules increases obviously with the increase in molecule carbon number. The largest deviation can be found in C40 isomers, the C40Mid is 0.188 GPa and C40End is 0.241 GPa, which means the Mid structure is 22.0% lower than the End molecules. In C30 and C40, the shear stresses of Iso and Mid are similar.
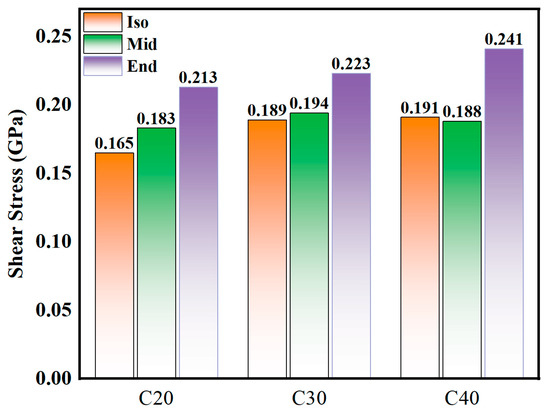
Figure 4.
Average shear stress of C20, C30, and C40 isomers.
3.2. Compression Distance and Molecular Volume
Figure 5 shows the compression distance of the simulated molecules at 500 MPa, which represents the lubricant film thickness variation before and after compression. It is found that the compression distance increases gradually as the molecular carbon number increases when the initial density of each model is identical. As for each molecule, Iso has the smallest amount of compression, and End has the largest. The smallest compression value is shown in C20Iso, which is 6.78 Å, the highest value is C40End, which is 11.26 Å, and the deviation between them can be up to 39.8%.
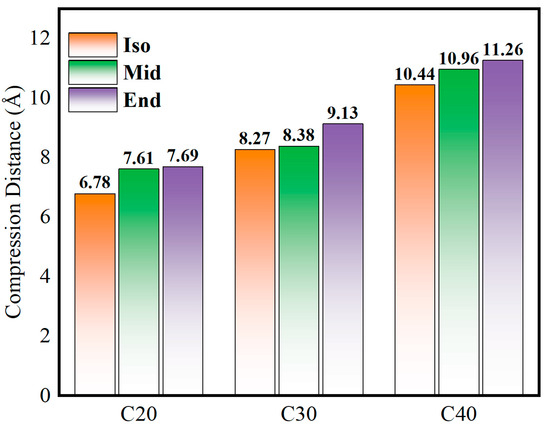
Figure 5.
Compression distance of C20, C30, and C40 isomers.
In Figure 6, the influence of compression distance is analyzed in conjunction with the molecular volume (vdW volume) and ellipsoidal volume. The volumes were obtained after molecule geometry optimization, which was calculated with a quantitative structure-activity relationship (QSAR) model. The molecular volume (vdW volume) is the volume of space in which the molecule is considered to be composed of spheres of atoms, which is calculated based on the van der Waals radius. The van der Waals radius is the effective radius that an atom exhibits in a non-bonded state and reflects the extent of space occupied by the atom in the molecule. Ellipsoidal volume is a geometric parameter used to describe the volume occupied by a molecule in three dimensions. It calculates the volume of a molecule by approximating it as an ellipsoid, thus reflecting the overall shape and size of the molecule. Unlike molecular volume, ellipsoidal volume contains the empty space accessible among the molecule’s carbon chains. In this study, the volume of molecules in Figure 1 was calculated after molecule geometry optimization, as these were used to build the lubricant layer in software, which influences the initial thickness of the lubricant film.
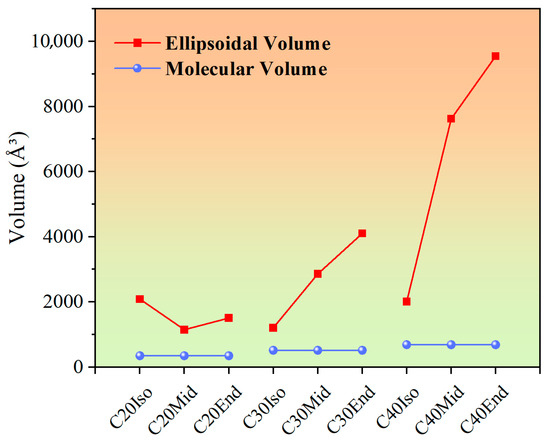
Figure 6.
Molecular volume (vdW volume) and ellipsoidal volume of C20, C30, and C40 isomers after geometry optimization.
In Figure 6, as the molecular carbon number increases, the molecular volume of C20, C30, and C40 increases gradually, which are approximately 348 Å3, 517 Å3, and 686 Å3. The slope of the ellipsoidal volume increases more sharply than the Molecular volume. This leads to an increase in empty space accessible to the intramolecular carbon chains, and when compressed, the increased gaps make it easier for the C30 and C40 molecules to be compressed. According to the previous PAO mixture simulation results, the PAO molecules with short side chains exhibited a rigid structure, while the molecules with long side chains exhibited the opposite and the tendency to orient along the shearing direction [30]. The molecules of C20 show shorter chain length compared with other structures, which makes them show little compression distance.
4. Discussion
4.1. Effect of Adsorption Energy
Figure 7 shows the adsorption energy of the oil film on the metal surface. The calculation formula is as follows:
where Etotal is the total energy of the system after compression, Esurface is the energy of an isolated metal surface, and Elub is the energy of an isolated organic molecule. The adsorption energy is usually negative. In this study, the adsorption energy is calculated with the absolute value. The higher the value, the stronger the adsorption.
Eads = |Etotal − (Esurface + Elub)|
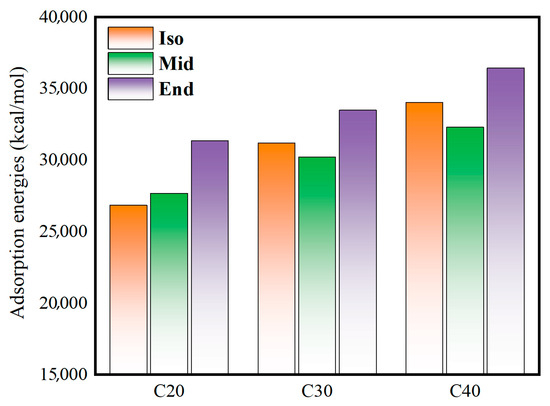
Figure 7.
Adsorption energies of oil films with metal walls.
Combined with the analysis of the shear stress in Figure 6, it can be found that the C20End, C30End, and C40End molecules with high adsorption energy have high shear stress. This indicates that the adsorption of molecules on the metal wall causes conformational changes in the molecules, which increases the internal friction between the molecules in the intermediate fluid layer and the adsorption layer and increases the overall shear stress.
The length of the main chain in the molecular structure of C20End, C30End, and C40End is longer than other structures, and the length of the branched chain in the molecular structure of C30Iso and C40Iso is longer; these long carbon chains make it easier to form an adsorption layer by adsorption on the metal wall [31].
Figure 8 shows the density distribution of the lubricant film. Generally, the density of each molecule is much higher near the metal surface compared with the middle region. The high adsorption energy of the End structure leads to a higher density near the metal surface. The density profiles in the middle region of C20 and C30 are very close. However, the density of C40 in the middle region shows that C40Iso shows the lowest density, and C40End shows the highest value, which is caused by the largest compression distance of C40End. The high compression distance leads to a compact lubricant layer and high density, which also increases the interaction among molecule branches and increases the shear stress.
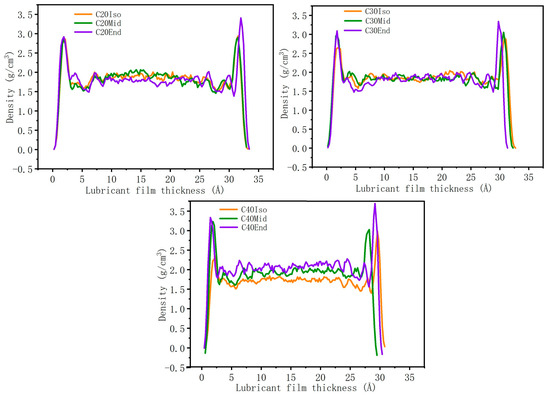
Figure 8.
Density distribution of lubricant film.
Wall slip is an important factor that influences the tribology property, which reduces the total shear stress. As shown in Figure 9, all the velocity profiles show an almost linear increasing trend, the reduced velocity gradient near the metal wall shows clear evidence of an adsorption layer, and there is no evidence of velocity profile slip at the iron-PAO interface, which means that the molecules do not show clear wall slip in the simulation condition. This may be caused by the relatively thin film thickness (~4 nm) and the high pressure, which constrained the molecules in the lubricant film and increased the interaction among molecule carbon branches.
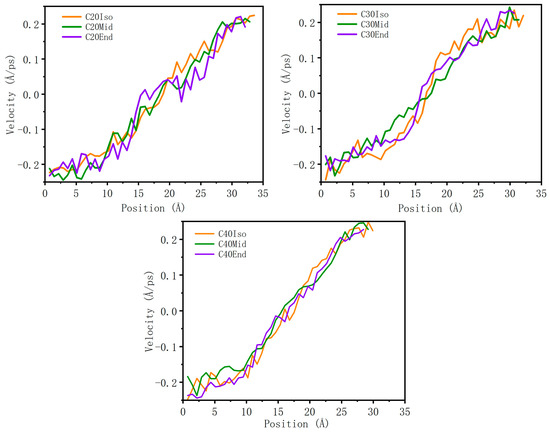
Figure 9.
Velocity profile of lubricant film.
4.2. Radius of Gyration
Figure 10 shows the radius of gyration of each molecule in free condition, which reflects the average distance of a molecule relative to the mass center and is a measure of the compactness or ductility of the molecule. The radius of gyration gradually increases as the carbon number of the molecule increases. Among the molecules, the Iso type has the smallest radius of gyration, and the Mid and End structures increase gradually. With the increase in molecular carbon number, the tendency to increase the radius of gyration of C30Mid-End and C40Mid-End is more obvious than that of C20Mid-End. This is also evidence that Iso molecules are more compact than End structures, and End structures are looser with more space within the molecule. Among simulated structures, the Iso structure has the least compression distance, and the End structure has the largest compression distance, which complies well with the variation in radius gyration. Therefore, in addition to the internal spatial factors of the molecules analyzed in Figure 6, the radius of gyration is another factor affecting the compressibility of the molecules.
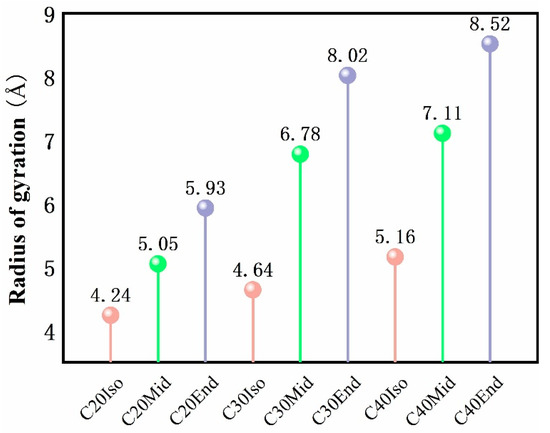
Figure 10.
Radius of gyration of each simulated molecule under free conditions. The color of pink stand for Iso structure, green stand for Mid structure, and the grey stand for End structure.
4.3. Radius of Gyration Evolution
Figure 11 shows the evolution in radius of gyration during the shear simulation. The radius of gyration may change due to chain segment orientation or untangling. Polymers have a reduced radius of gyration, indicating that the molecules are curled up during friction, which may reduce the frictional resistance among molecules. If the radius of gyration increases, it indicates that the shear effect stretches the molecular chain, and the local entanglement and pulling between the molecular carbon chains are obvious, which increases the local internal friction.
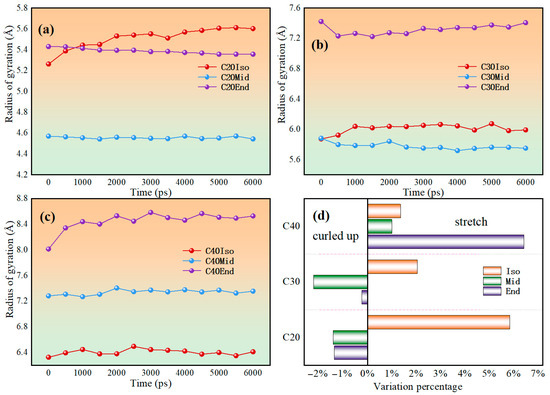
Figure 11.
Radius gyration evolution of (a) C20 molecules, (b) C30 molecules, (c) C40 molecules, and (d) variation percentage of each lubricant model during the shear process.
The simulation results show that only C20Mid, C20End, C30Mid, and C30End decrease with shear; other molecules show an increased trend. This indicates that during shear, most of the molecules have a gradual increase in pulling of the carbon chain, while the entanglement of carbon chains in C20Mid, C20End, C30Mid, and C30End decreases. This means the C20Mid, C20End, C30Mid, and C30End have a reduced friction force during shear, while other molecules increase gradually.
4.4. Mean Square Displacement
The mean square displacement reflects the diffusion of lubricant molecules over time and the flexibility of the molecular carbon chains. If the value is larger, it may reflect that the molecules have a wider range of motion, the molecular carbon chains are more flexible, and the molecules are less entangled with each other, which reduces friction.
Figure 12 shows the mean square displacement curve during confined shear, and Figure 13 shows the diffusion coefficient of each molecule. Generally, the mean square displacement of C30 is the largest. Among different structures, the Mid structure of C20, C30, and C40 is the lowest. This means the Iso structure has less entanglement among branched chains, which have less internal friction among molecules, and this matches well with the results of shear stress. As for the Iso structure, C20 is a linear molecule, and C30 and C40 are molecules with multi-branched chains, showing significant differences. As for C20, the mean square displacement of the Iso structure is higher than the mid and end structures, while the shear stress is lower compared with the other structures. This means the small linear molecules are flexible to move and exhibit low internal friction [32]. As for the End structure, C30 is the highest, and C40 and C20 are gradually lower. The End structures have carbon branches near the end of the main chain, which makes them more easily entangled with molecules near the side, resulting in an increase in shear stress. C40 has a further increase in carbon number and carbon chain length, but the mean square displacement is reduced compared with C30, which is related to the characteristics of the oil film. The molecules close to the metal wall are adsorbed on the metal surface, and the larger molecular size of C40 implies that more atoms are constrained on the metal surface, and the motion is limited.
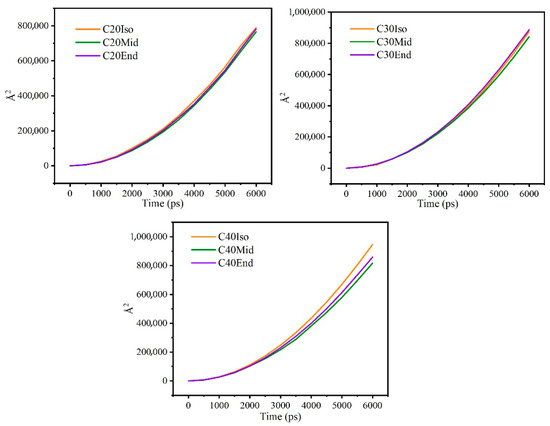
Figure 12.
Mean square displacement curve during confined shear.
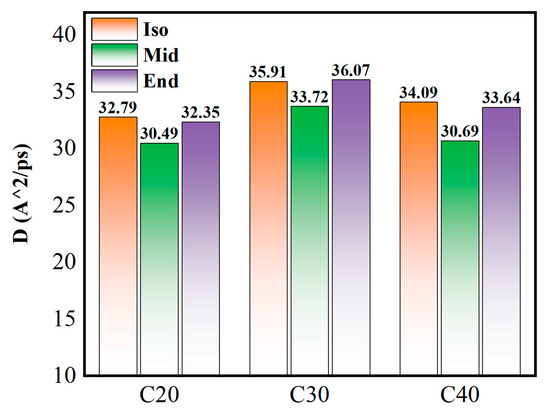
Figure 13.
Diffusion coefficient of each molecule.
5. Conclusions
In this paper, the molecular dynamics simulation was applied to study the lubrication performance of base oils with different molecular structure characteristics. Comparison studies were carried out under the same conditions by using the iron-oil-iron model, and the following conclusions were obtained.
(1) Among the various molecular structure characteristics, the shear stresses of Iso and Mid are smaller than End structure. The largest deviation can be found in C40Mid (0.188 GPa) and C40End (0.241 GPa), which means the Mid structure is 22.0% lower than the End molecules. This indicates that the molecules of the Mid type have better friction reduction performance. The compression of the Iso structure is the smallest, and the Mid and End gradually increase. The smallest compression value is shown in C20Iso, which is 6.78 Å, and the highest value is C40End, which is 11.26 Å, the deviation between which can be up to 39.8%. This indicates that the molecules of the Iso structure have the largest oil film thickness and the best load-bearing performance. Among the nine molecules, C20Iso has the smallest compression distance of 6.78 Å and a low shear stress of 0.165 GPa, which shows excellent performance both in load-bearing and friction reduction.
(2) In the case of the same structure type, with the increase in molecular carbon number, the adsorption energy between oil molecules and the iron wall surface increases, the radius of gyration of most molecules increases gradually with shear, and the shear stress of molecules increases and the compression increases. This indicates that the macroscopic friction increases, the oil film thickness decreases, and the lubrication performance decreases.
(3) In the case of the same number of molecular carbon, with the molecular structure changes, End’s radius of gyration is the largest, Iso’s radius of gyration is the smallest, proving that End’s molecules are more loose, Iso’s molecules are more compact, combined with End’s molecules with a larger internal space, therefore the End’s compression is larger and Iso’s compression is smaller; The adsorption energy of End is the highest and the adsorption energy of Mid is the smallest, and the adsorption of molecules on the metal wall produces a conformational change in the molecules, which increases the internal friction between the molecules in the intermediate fluid layer and the adsorption layer, resulting in the maximum shear stress of End and the minimum shear stress of Mid.
Author Contributions
Conceptualization and writing—original draft preparation, B.T.; methodology and investigation, Y.S. and F.Z.; project administration and funding acquisition, W.W.; software and data curation, C.H., T.Z. and J.L.; writing—review and editing, H.X. and C.C.; visualization and funding acquisition, H.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China, grant number (52201383); the National Key Laboratory of Marine Engine Science and Technology, grant number (LAB-2025-12-WD); the Natural Science Foundation of Shandong Province, grant numbers (ZR2023ME067); and the Basic Research Project of Yantai Science and Technology Innovation Development Plan, grant number (2023JCYJ054).
Data Availability Statement
All data generated during this study are included in this article, and the datasets are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Taylor, R.I. Tribology and energy efficiency: From molecules to lubricated contacts to complete machines. Faraday Discuss. 2012, 156, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Sarpal, A.S.; Sastry, M.I.S.; Bansal, V.; Singh, I.; Mazumdar, S.K.; Basu, B. Correlation of structure and properties of groups I to III base oils. Lubr. Sci. 2012, 24, 199–215. [Google Scholar] [CrossRef]
- Zhang, X.A.; Zhao, Y.Z.; Ma, K.; Wang, Q. Friction behavior and wear protection ability of selected base lubricants. Friction 2016, 4, 72–83. [Google Scholar] [CrossRef]
- Airey, J.; Spencer, M.; Greenwood, R.; Simmons, M. The effect of gas turbine lubricant base oil molecular structure on friction. Tribol. Int. 2020, 146, 106052. [Google Scholar] [CrossRef]
- Mirzaamiri, R.; Akbarzadeh, S.; Ziaei-Rad, S.; Shin, D.G.; Kim, D.E. Molecular dynamics simulation and experimental investigation of tribological behavior of nanodiamonds in aqueous suspensions. Tribol. Int. 2021, 156, 106838. [Google Scholar] [CrossRef]
- Bucholz, E.W.; Phillpot, S.R.; Sinnott, S.B. Molecular dynamics investigation of the lubrication mechanism of carbon nano-onions. Comput. Mater. Sci. 2012, 54, 91–96. [Google Scholar] [CrossRef]
- Washizu, H.; Ohmori, T. Molecular dynamics simulations of elastohydrodynamic lubrication oil film. Lubr. Sci. 2010, 22, 323–340. [Google Scholar] [CrossRef]
- Stephan, S.; Schmitt, S.; Hasse, H.; Urbassek, H.M. Molecular dynamics simulation of the Stribeck curve: Boundary lubrication, mixed lubrication, and hydrodynamic lubrication on the atomistic level. Friction 2023, 11, 2342–2366. [Google Scholar] [CrossRef]
- Cheng, Q.H.; Zheng, T.; Yang, G.; Zhang, H.C. Molecular Dynamics Simulation of Loading and Lubrication Mechanisms of Konjac Glucomannan Hydration Layer. Acta Polym. Sin. 2023, 54, 1784–1794. [Google Scholar]
- Xue, J.Y.; Dong, S.Q.; Mi, P.K.; Wang, L.B.; Wang, S.H.; Zhang, Z.; Zhang, Z.G.; Hu, J.S. Study of the structure-activity relationship of metallocene-catalysed poly-α-olefin (mPAO) base oil. Mol. Syst. Des. Eng. 2021, 6, 722–729. [Google Scholar] [CrossRef]
- Du, R.; Zhang, A.Y.; Du, Z.H.; Zhang, X.Y. Molecular Dynamics Simulation on Thin-Film Lubrication of a Mixture of Three Alkanes. Materials 2020, 13, 3689. [Google Scholar] [CrossRef] [PubMed]
- Mercier Franco, L.F.; Firoozabadi, A. Computation of Shear Viscosity by a Consistent Method in Equilibrium Molecular Dynamics Simulations: Applications to 1-Decene Oligomers. J. Phys. Chem. B 2023, 127, 10043–10051. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Li, H.; Lu, Y.; Sun, L.; Xu, S.F.; Fan, X.L. Synergistic lubrication of organic friction modifiers in boundary lubrication regime by molecular dynamics simulations. Appl. Surf. Sci. 2023, 623, 157087. [Google Scholar] [CrossRef]
- Zheng, X.; Su, L.H.; Deng, G.Y.; Zhang, J.; Zhu, H.T.; Tieu, A.K. Study on Lubrication Characteristics of C4-Alkane and Nanoparticle during Boundary Friction by Molecular Dynamics Simulation. Metals 2021, 11, 1464. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, H.; Lu, S.P.; Chen, Y.H. Effect of Pressure on Boundary Slip of Thin Film Lubrication Using Atomistic Simulation. J. Wuhan Univ. Technol.-Mater. Sci. Ed. 2020, 35, 47–52. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, H.T.; Kosasih, B.; Tieu, A.K. A molecular dynamics simulation of boundary lubrication: The effect of n-alkanes chain length and normal load. Wear 2013, 301, 62–69. [Google Scholar] [CrossRef]
- Katsukawa, R.; Van Sang, L.; Tomiyama, E.; Washizu, H. High-Pressure Lubrication of Polyethylethylene by Molecular Dynamics Approach. Tribol. Lett. 2022, 70, 101. [Google Scholar] [CrossRef]
- Mehrnia, S.; Pelz, P.F. Tribological Design by Molecular Dynamics Simulation: Influence of the Molecular Structure on Wall Slip and Bulk Shear. Chem. Eng. Technol. 2022, 46, 95–101. [Google Scholar] [CrossRef]
- Hou, X.J.; An, H.; Ma, Y.X.; Chu, C.; Ali, M.K.A. The effect of the number of branched hydrocarbon molecules on boundary lubrication of ZnO nanofluids by using molecular dynamics simulation. Mater. Today Commun. 2024, 38, 108585. [Google Scholar] [CrossRef]
- Xu, Q.; Tang, X.; Zhang, J.; Hu, Y.Z.; Ma, T.B. Unraveling Tribochemistry and Self-Lubrication Mechanism of Polytetrafluoroethylene by Reactive Coarse-Grained Molecular Dynamics Simulations. ACS Appl. Mater. Interfaces 2023, 15, 45506–45515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.X.; Lu, B.; Wu, Y.H.; Wang, J.H.; Zhang, X.Y.; Wang, L.Y. Molecular dynamics simulation and experimental study on the lubrication of graphene additive films. Proc. Inst. Mech. Eng. Part J J. Eng. Tribol. 2023, 234, 1957–1972. [Google Scholar] [CrossRef]
- Savio, D.; Fillot, N.; Vergne, P.A. A Molecular Dynamics Study of the Transition from Ultra-Thin Film Lubrication Toward Local Film Breakdown. Tribol. Lett. 2013, 50, 207–220. [Google Scholar] [CrossRef]
- Xiong, S.; Zhang, X.M.; Liang, D. Molecular dynamics simulation of friction coefficient of Fe-Al during lubrication. Comput. Mater. Sci. 2023, 217, 111895. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications–Overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular-Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, M.; Liu, J.; Liu, G.; Zhou, F. Molecular dynamics simulations of adsorption behavior of organic friction modifiers on hydrophilic silica surfaces under the effects of surface coverage and contact pressure. Tribol. Int. 2021, 156, 106826. [Google Scholar] [CrossRef]
- Du, J.; Jin, Y.; Hou, S.; Jin, R.; Wang, Q. Effect of component characteristics on mechanical properties of asphalt: A molecular dynamics study. Case Stud. Constr. Mater. 2023, 18, e02007. [Google Scholar] [CrossRef]
- Tang, L.; Li, Y.; He, E.; Hao, M.; Ren, S. Molecular simulation of tribology behavior of nano ZnO/nitrile-butadiene rubber composites. Acta Mater. Compos. Sin. 2020, 37, 690–695. [Google Scholar]
- Wang, W.; Zhang, X.; Li, Y.; Huang, R.; Xu, J.; Yang, L. Effects of molecular structures of poly α-olefin mixture on nano-scale thin film lubrication. Mater. Today Commun. 2020, 25, 101500. [Google Scholar] [CrossRef]
- Turgut, C.; Pandiyan, S.; Mether, L.; Belmahi, M.; Nordlund, K.; Philipp, P. Influence of alkane chain length on adsorption on an α-alumina surface by MD simulations. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2015, 352, 206–209. [Google Scholar] [CrossRef]
- Ewen, J.P.; Gao, H.; Müser, M.H.; Dini, D. Shear heating, flow, and friction of confined molecular fluids at high pressure. Phys. Chem. Chem. Phys. 2019, 21, 5813. [Google Scholar] [CrossRef] [PubMed]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).