History-Dependent Stress Relaxation of Liquids under High-Confinement: A Molecular Dynamics Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
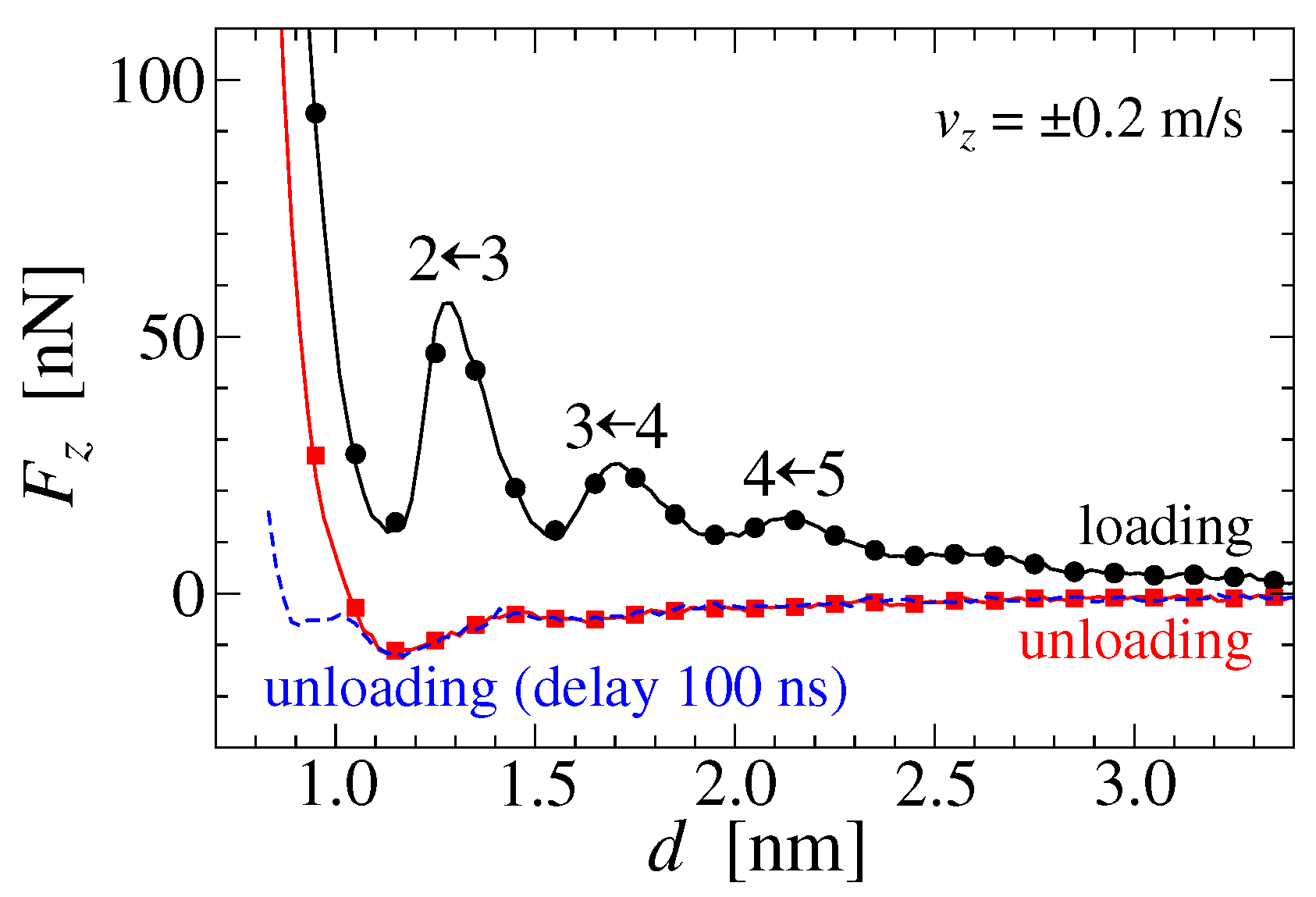
3.1. Solvation Force
3.2. Stress Relaxation
3.3. Radius of Gyration
3.4. Mean-Squared Displacement
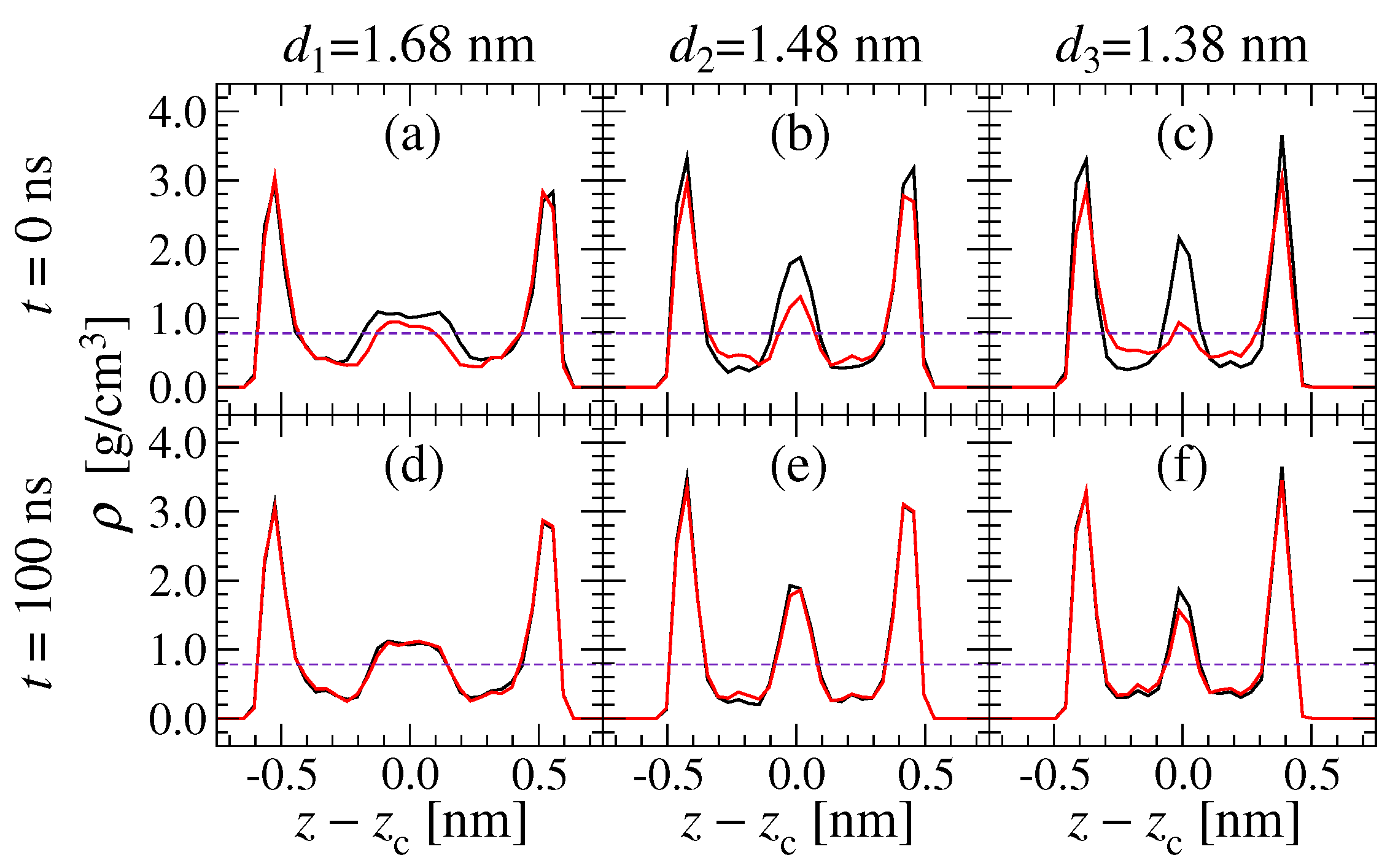
3.5. Density Profile
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Simon, P.; Gogotsi, Y. Materials for electrochemical capacitors. Nat. Mater. 2008, 7, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [Green Version]
- Rasaiah, J.C.; Garde, S.; Hummer, G. Water in Nonpolar Confinement: From Nanotubes to Proteins and Beyond. Annu. Rev. Phys. Chem. 2008, 59, 713–740. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.L.; McKenna, G.B. Vitrification and Crystallization of Organic Liquids Confined to Nanoscale Pores. Chem. Mater. 1996, 8, 2128–2137. [Google Scholar] [CrossRef]
- Horn, R.G.; Israelachvili, J.N. Direct measurement of structural forces between two surfaces in a nonpolar liquid. J. Chem. Phys. 1981, 75, 1400–1411. [Google Scholar] [CrossRef]
- Ballamudi, R.K.; Bitsanis, I.A. Energetically driven liquid–solid transitions in molecularly thin n-octane films. J. Chem. Phys. 1996, 105, 7774–7782. [Google Scholar] [CrossRef]
- Van Alsten, J.; Granick, S. Molecular Tribometry of Ultrathin Liquid Films. Phys. Rev. Lett. 1988, 61, 2570–2573. [Google Scholar] [CrossRef]
- Granick, S. Motions and Relaxations of Confined Liquids. Science 1991, 253, 1374–1379. [Google Scholar] [CrossRef] [Green Version]
- Sivebaek, I.M.; Samoilov, V.N.; Persson, B.N.J. Effective Viscosity of Confined Hydrocarbons. Phys. Rev. Lett. 2012, 108, 036102. [Google Scholar] [CrossRef] [Green Version]
- Christenson, H.K. Experimental measurements of solvation forces in nonpolar liquids. J. Chem. Phys. 1983, 78, 6906–6913. [Google Scholar] [CrossRef]
- Krass, M.D.; Krämer, G.; Dellwo, U.; Bennewitz, R. Molecular Layering in Nanometer-Confined Lubricants. Tribol. Lett. 2018, 66, 87. [Google Scholar] [CrossRef]
- Nygård, K. Local structure and density fluctuations in confined fluids. Curr. Opin. Colloid Interface Sci. 2016, 22, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Nygård, K.; Sarman, S.; Hyltegren, K.; Chodankar, S.; Perret, E.; Buitenhuis, J.; van der Veen, J.F.; Kjellander, R. Density Fluctuations of Hard-Sphere Fluids in Narrow Confinement. Phys. Rev. X 2016, 6, 011014. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.E.; Wiodm, B. Decay of Correlations in Linear Systems. J. Chem. Phys. 1969, 50, 3756–3772. [Google Scholar] [CrossRef]
- Kjellander, R.; Sarman, S. Pair correlations of non-uniform hard-sphere fluids in narrow slits and the mechanism of oscillatory solvation forces. J. Chem. Soc. Faraday Trans. 1991, 87, 1869–1881. [Google Scholar] [CrossRef]
- Krass, M.D.; Gosvami, N.N.; Carpick, R.W.; Müser, M.H.; Bennewitz, R. Dynamic shear force microscopy of viscosity in nanometer-confined hexadecane layers. J. Phys.-Condens. Matter. 2016, 28, 134004. [Google Scholar] [CrossRef]
- Gao, H.; Müser, M.H. Why liquids can appear to solidify during squeeze-out–Even when they don’t. J. Colloid Interface Sci. 2020, 562, 273–278. [Google Scholar] [CrossRef]
- Rouse, P.E. A Theory of the Linear Viscoelastic Properties of Dilute Solutions of Coiling Polymers. J. Chem. Phys. 1953, 21, 1272–1280. [Google Scholar] [CrossRef]
- Hu, H.W.; Carson, G.A.; Granick, S. Relaxation time of confined liquids under shear. Phys. Rev. Lett. 1991, 66, 2758–2761. [Google Scholar] [CrossRef]
- Granick, S.; Hu, H.W. Nanorheology of Confined Polymer Melts. 1. Linear Shear Response at Strongly Adsorbing Surfaces. Langmuir 1994, 10, 3857–3866. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Adams, G.E. Measurement of forces between two mica surfaces in aqueous electrolyte solutions in the range 0–100 nm. J. Chem. Soc. Faraday Trans. 1978, 74, 975–1001. [Google Scholar] [CrossRef]
- Cui, S.T.; Cummings, P.T.; Cochran, H.D. Molecular simulation of the transition from liquidlike to solidlike behavior in complex fluids confined to nanoscale gaps. J. Chem. Phys. 2001, 114, 7189–7195. [Google Scholar] [CrossRef]
- Alba-Simionesco, C.; Coasne, B.; Dosseh, G.; Dudziak, G.; Gubbins, K.E.; Radhakrishnan, R.; Sliwinska-Bartkowiak, M. Effects of confinement on freezing and melting. J. Phys.-Condens. Matter. 2006, 18, R15–R68. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Kumacheva, E. Confinement-Induced Phase Transitions in Simple Liquids. Science 1995, 269, 816–819. [Google Scholar] [CrossRef]
- Kienle, D.F.; Kuhl, T.L. Density and Phase State of a Confined Nonpolar Fluid. Phys. Rev. Lett. 2016, 117, 036101. [Google Scholar] [CrossRef] [Green Version]
- Atkin, R.; Warr, G.G. Structure in Confined Room-Temperature Ionic Liquids. J. Phys. Chem. C 2007, 111, 5162–5168. [Google Scholar] [CrossRef]
- Bureau, L. Rate Effects on Layering of a Confined Linear Alkane. Phys. Rev. Lett. 2007, 99, 225503. [Google Scholar] [CrossRef] [Green Version]
- Müser, M.H.; Robbins, M.O. Conditions for static friction between flat crystalline surfaces. Phys. Rev. B 2000, 61, 2335–2342. [Google Scholar] [CrossRef]
- Stevens, M.J.; Mondello, M.; Grest, G.S.; Cui, S.T.; Cochran, H.D.; Cummings, P.T. Comparison of shear flow of hexadecane in a confined geometry and in bulk. J. Chem. Phys. 1997, 106, 7303–7314. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, S.J.; Welland, M.E.; Rayment, T. Solvation forces near a graphite surface measured with an atomic force microscope. Appl. Phys. Lett. 1992, 60, 2356–2358. [Google Scholar] [CrossRef]
- Heuberger, M.; Zäch, M. Nanofluidics: Structural Forces, Density Anomalies, and the Pivotal Role of Nanoparticles. Langmuir 2003, 19, 1943–1947. [Google Scholar] [CrossRef]
- Gee, M.L.; McGuiggan, P.M.; Israelachvili, J.N.; Homola, A.M. Liquid to solidlike transitions of molecularly thin films under shear. J. Chem. Phys. 1990, 93, 1895–1906. [Google Scholar] [CrossRef]
- Carson, G.; Hu, H.W.; Granick, S. Molecular Tribology of Fluid Lubrication: Shear Thinning. Tribol. Trans. 1992, 35, 405–410. [Google Scholar] [CrossRef]
- Leng, Y.; Cummings, P.T. Hydration structure of water confined between mica surfaces. J. Chem. Phys. 2006, 124, 074711. [Google Scholar] [CrossRef]
- Wang, J.C.; Fichthorn, K.A. A method for molecular dynamics simulation of confined fluids. J. Chem. Phys. 2000, 112, 8252–8259. [Google Scholar] [CrossRef]
- Wang, Y.; Hill, K.; Harris, J.G. Confined thin films of a linear and branched octane. A comparison of the structure and solvation forces using molecular dynamics simulations. J. Chem. Phys. 1994, 100, 3276–3285. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Novel Configurational-Bias Monte Carlo Method for Branched Molecules. Transferable Potentials for Phase Equilibria. 2. United-Atom Description of Branched Alkanes. J. Phys. Chem. B 1999, 103, 4508–4517. [Google Scholar] [CrossRef]
- Wick, C.D.; Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 4. United-Atom Description of Linear and Branched Alkenes and Alkylbenzenes. J. Phys. Chem. B 2000, 104, 8008–8016. [Google Scholar] [CrossRef] [Green Version]
- Ewen, J.P.; Gao, H.; Müser, M.H.; Dini, D. Shear heating, flow, and friction of confined molecular fluids at high pressure. Phys. Chem. Chem. Phys. 2019, 21, 5813–5823. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Price, M.L.P.; Ostrovsky, D.; Jorgensen, W.L. Gas-phase and liquid-state properties of esters, nitriles, and nitro compounds with the OPLS-AA force field. J. Comput. Chem. 2001, 22, 1340–1352. [Google Scholar] [CrossRef]
- Lorentz, H.A. Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, D. Sur le melange des gaz. Comptes. Rendus. Acad. Sci. 1898, 126, 1703–1855. [Google Scholar]
- Savio, D.; Fillot, N.; Vergne, P.; Zaccheddu, M. A Model for Wall Slip Prediction of Confined n-Alkanes: Effect of Wall-Fluid Interaction Versus Fluid Resistance. Tribol. Lett. 2012, 46, 1573–2711. [Google Scholar] [CrossRef]
- Gao, H.; Ewen, J.P.; Hartkamp, R.; Müser, M.H.; Dini, D. Scale-Dependent Friction–Coverage Relations and Nonlocal Dissipation in Surfactant Monolayers. Langmuir 2021, 37, 2406–2418. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Hoth, J.; Hausen, F.; Müser, M.H.; Bennewitz, R. Force microscopy of layering and friction in an ionic liquid. J. Phys.-Condens. Matter. 2014, 26, 284110. [Google Scholar] [CrossRef]
- Tsai, D.H. The virial theorem and stress calculation in molecular dynamics. J. Chem. Phys. 1979, 70, 1375–1382. [Google Scholar] [CrossRef]
- Zhou, M. A new look at the atomic level virial stress: On continuum-molecular system equivalence. Proc. R. Soc. London. Ser. A Math. Phys. Eng. Sci. 2003, 459, 2347–2392. [Google Scholar] [CrossRef]
- Todd, B.D.; Evans, D.J.; Daivis, P.J. Pressure tensor for inhomogeneous fluids. Phys. Rev. E 1995, 52, 1627–1638. [Google Scholar] [CrossRef]
- Hartkamp, R.; Ghosh, A.; Weinhart, T.; Luding, S. A study of the anisotropy of stress in a fluid confined in a nanochannel. J. Chem. Phys. 2012, 137, 044711. [Google Scholar] [CrossRef]
- Mukherji, D.; Müser, M.H. Possible explanation of the Λ-shape anomaly in polymer surface diffusion. Phys. Rev. E 2006, 74, 010601. [Google Scholar] [CrossRef]
- Shewmon, P. High Diffusivity Paths. In Diffusion in Solids; Springer: Cham, Switzerland, 2016; pp. 189–222. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H. History-Dependent Stress Relaxation of Liquids under High-Confinement: A Molecular Dynamics Study. Lubricants 2022, 10, 15. https://doi.org/10.3390/lubricants10020015
Gao H. History-Dependent Stress Relaxation of Liquids under High-Confinement: A Molecular Dynamics Study. Lubricants. 2022; 10(2):15. https://doi.org/10.3390/lubricants10020015
Chicago/Turabian StyleGao, Hongyu. 2022. "History-Dependent Stress Relaxation of Liquids under High-Confinement: A Molecular Dynamics Study" Lubricants 10, no. 2: 15. https://doi.org/10.3390/lubricants10020015
APA StyleGao, H. (2022). History-Dependent Stress Relaxation of Liquids under High-Confinement: A Molecular Dynamics Study. Lubricants, 10(2), 15. https://doi.org/10.3390/lubricants10020015