The Astrochemistry Implications of Quantum Chemical Normal Modes Vibrational Analysis



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Brief Summary of Our Previous Work
- In hydrocarbons within the realm of classical molecular structure only C-H stretching vibrations occur. This holds for any combination of different aromatic (sp), olefinic (sp) and aliphatic (sp) groups within a molecule. Considering the low amplitude motions in our analysis we did not find the signatures of other types of vibrations in this spectral region.
- For the above types of molecules the aromatic (sp) C-H vibrations do not show any common wavelength coverage with aliphatic (sp) stretching modes (non-overlap wavelength range). The vibrational motions of these two types of C-H bonds are reported to be uncoupled.
- Olefinic (sp) C-H stretching shows a common wavelength coverage with aromatic (sp) C-H vibrations (overlapped wavelength ranges ). However, vibrational motions of these two bond types occur independently without any couplings.
- Olefinic (sp) C-H stretching motion couples partly with aliphatic (sp) C-H vibrations.
- These vibrational characteristics of olefinic C-H stretching motions can glue the wavelength range of aliphatic (sp) and aromatic (sp) C-H stretching vibrations. In other words, they can make an indirect coupling between these two uncoupled and non-overlapped vibrational motions. This effect can be considered as one of the origins of the formation of a plateau in this spectral region.
- The symmetric and asymmetric C-H stretching motions of methyl and methylene groups are highly coupled with common wavelength coverage of the resultant features. Thus, vibrational motions of these two functional groups in this wavelength region are difficult to discriminate against under different UIE features.
- In neutral honeycomb PAH molecules, the stretching vibrations of C-H bonds in the bay and non-bay positions show considerable coupling in addition to their common wavelength coverage.
3. New Results from This Work
3.1. The 3 m Region
Models: PAH, PAHs with Side Groups, Aliphatic Hydrocarbons and MAONs
3.2. The 6–10 m Region
Model Used: PAH
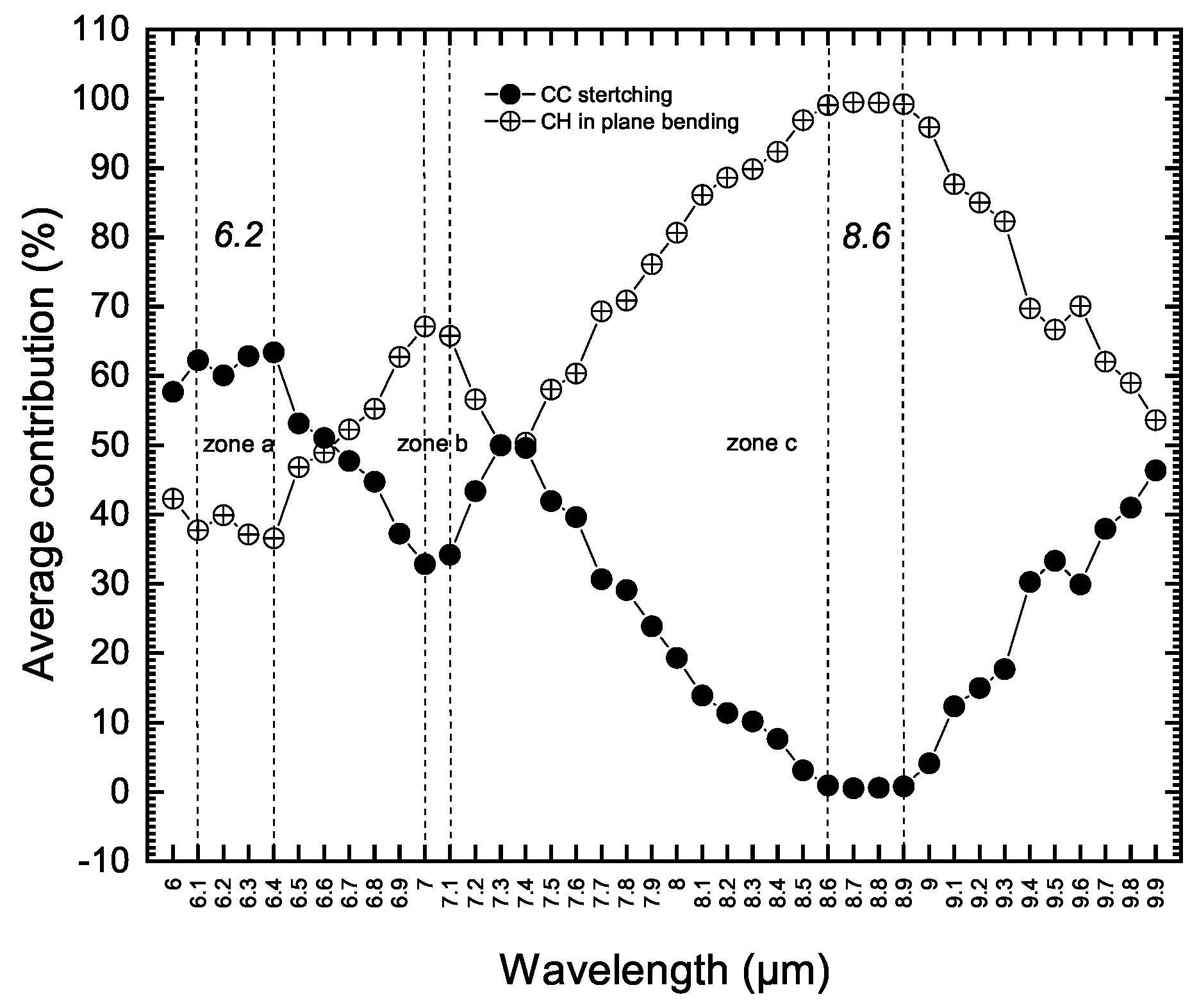
- All normal-mode vibrations (IR and Raman active modes) within this wavelength range occur in the symmetry plane of the PAH molecule. They are all in-plane modes. We did not detect any low amplitude out of plane modes through our quantitative analysis.
- All vibrations are reported as highly coupled modes. This includes the 6.2 m feature described as an uncoupled C-C stretching band (zone (a) in Figure 2). Only the wavelength range of 8.6 to 8.9 m is found to contain pure aromatic C-H in-plane bending modes (zone (c) in Figure 2).These pictures of vibrations, derived and assigned purely from theory, are in very good agreement with the results of experimental spectroscopy reported by Jobelin et al. [29] from IR spectra of numbers of PAH molecules. This agreement with experimental work demonstrates the reliability of our quantitative vibrational analysis methodology which is a key point to exploring the origin of UIE bands at longer wavelengths.
- In Figure 2 we have labeled and described these vibrations as coupled C-C stretching and C-H in-plane modes. These are commonly used labels. These types of motions also occur at longer wavelengths in PAH molecules. We explain later a possible way of discriminating such vibrations and assigning them to intrinsic fragments. It should be mentioned that the comprehensive vibrational analysis of this 6–10 m region and the 3 m part of the spectra has led us to suggest that the olefinic carrier of the 6 m UIE feature observed in the famous protoplanetary red rectangle nebulae HD44179 [22]. This feature was previously assigned to CO molecule.
3.3. The 11–15 m Region
Mode Usesl: PAH
3.4. Skeletal Modes
Model Used: MAONs
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Duley, W.W.; Williams, D.A. The infrared spectrum of interstellar dust: Surface functional groups on carbon. Mon. Not. R. Astron. Soc. 1981, 196, 269–274. [Google Scholar] [CrossRef]
- Allamandola, L.J.; Tielens, A.G.G.M.; Barker, J.R. Polycyclic aromatic hydrocarbons and the unidentified infrared emission bands—Auto exhaust along the Milky Way. Astrophys. J. 1985, 290, L25–L28. [Google Scholar] [CrossRef]
- Wagner, D.R.; Kim, H.S.; Saykally, R.J. Peripherally Hydrogenated Neutral Polycyclic Aromatic Hydrocarbons as Carriers of the 3 Micron Interstellar Infrared Emission Complex: Results from Single-Photon Infrared Emission Spectroscopy. Astrophys. J. 2000, 545, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, D.M.; Allamandola, L.J. Polycyclic Aromatic Hydrocarbons and Infrared Astrophysics: The State of the PAH Model and a Possible Tracer of Nitrogen in Carbon-Rich Dust. Astrophys. Dust ASP Conf. Ser. 2004, 309, 665–668. [Google Scholar] [CrossRef]
- Duley, W.W.; Grishko, V.I. Evolution of Carbon Dust in Aromatic Infrared Emission Sources: Formation of Nanodiamonds. Astrophys. J. Lett. 2001, 554, L209–L212. [Google Scholar] [CrossRef]
- Cataldo, F.; Keheyan, Y.; Heymann, D. A new model for the interpretation of the unidentified infrared bands (UIBS) of the diffuse interstellar medium and of the protoplanetary nebulae. Int. J. Astrobiol. 2002, 1, 79–86. [Google Scholar] [CrossRef]
- Wada, S.; Tokunaga, A.T. Carbonaceous onino-like particles: A possible component of the interstellar medium. In Natural Fullerences and Related Structures of Elemental Carbon; Rietmeijer, F.J.M., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 31–52. ISBN 978-1-4020-4135-8. [Google Scholar]
- Kwok, S.; Zhang, Y. Mixed aromatic-aliphatic organic nanoparticles as carriers of unidentified infrared emission features. Nature 2011, 479, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Sadjadi, S.; Zhang, Y.; Kwok, S. A Theoretical Study on the Vibrational Spectra of Polycyclic Aromatic Hydrocarbon Molecules with Aliphatic Sidegroups. Astrophys. J. 2015, 801, 34. [Google Scholar] [CrossRef]
- Morino, Y.; Kuchitsu, K. A Note on the Classification of Normal Vibrations of Molecules. J. Chem. Phys. 1952, 20, 1809–1810. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A new way of analyzing vibrational spectra. I. Derivation of adiabatic internal modes. Int. J. Quantum Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. IV. Application and Testing of Adiabatic Modes within the Concept of the Characterization of Normal Modes. Int. J. Quantum Chem. 1998, 67, 41–55. [Google Scholar] [CrossRef]
- Mackie, C.J.; Candian, A.; Huang, X.; Maltseva, E.; Petrignani, A.; Oomens, J.; Buma, W.J.; Lee, T.J.; Tielens, A.G.G.M. The anharmonic quartic force field infrared spectra of three aromatic hydrocarbons: Naphthalene, anthracene and tetracene. J. Chem. Phys. 2015, 143, 224314(1)–224314(15). [Google Scholar] [CrossRef] [PubMed]
- Hanson-Heine, M.W.D.; George, M.W.; Besley, N.A. Investigating the Calculation of Anharmonic Vibrational Frequencies Using Force Fields Derived from Density Functional Theory. J. Phys. Chem. A 2012, 116, 4417–4425. [Google Scholar] [CrossRef] [PubMed]
- Miani, A.; Cané, E.; Palmieri, P.; Trombetti, A. Experimental and theoretical anharmonicity for benzene using density functional theory. J. Chem. Phys. 2000, 112, 248–259. [Google Scholar] [CrossRef]
- Guntram, R.; Tomica, H. A combined variational and perturbational study on the vibrational spectrum of P2F4. Chem. Phys. 2008, 346, 160–166. [Google Scholar] [CrossRef]
- Dunham, J.L. The energy levels of rotating vibrator. Phys. Rev. 1932, 41, 721–731. [Google Scholar] [CrossRef]
- Pavlyuchkoa, A.I.; Yurchenko, S.N.; Tennysonb, J. Hybrid variational–perturbation method for calculating ro-vibrational energy levels of polyatomic molecules. Mol. Phys. 2015, 113, 1559–1575. [Google Scholar] [CrossRef]
- Sadjadi, S.; Zhang, Y.; Kwok, S. On the Origin of the 3.3 μm Unidentified Infrared Emission Feature. Astrophys. J. 2017, 845, 123. [Google Scholar] [CrossRef]
- Tao, Y.; Tian, C.; Verma, N.; Zou, W.; Wang, C.; Cremer, D.; Kraka, E. Recovering Intrinsic Fragmental Vibrations Using the Generalized Subsytem Vibrational Analysis. J. Chem. Theory Comput. 2018, 14, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Sadjadi, S.; Kwok, S.; Zhang, Y. Theoretical infrared spectra of MAON molecules. J. Phys. Conf. Ser. 2016, 728, 062003. [Google Scholar] [CrossRef]
- Hsia, C.-H.; Sadjadi, S.; Zhang, Y.; Kwok, S. The 6 μm Feature as a Tracer of Aliphatic Components of Interstellar Carbonaceous Grains. Astrophys. J. 2016, 832, 213. [Google Scholar] [CrossRef]
- Sadjadi, S.; Zhang, Y.; Kwok, S. On the Origin of the 11.3 Micron Unidentified Infrared Emission Feature. Astrophys. J. 2015, 807, 95. [Google Scholar] [CrossRef]
- Hudgins, D.M.; Allamandola, L.J. Interstellar PAH Emission in the 11-14 Micron Region: New Insightsfrom Laboratory Data and a Tracer of Ionized PAHs. Astrophys. J. Lett. 1999, 516, L41–L44. [Google Scholar] [CrossRef]
- Laury, M.L.; Carlson, M.J.; Wilson, A.K. Vibrational frequency scale factors for density functional theory and the polarization consistent basis sets. J. Comput. Chem. 2012, 33, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Richard, F. Bader. In Atoms in Molecules A Quantum Theory; Oxford University: New York, NY, USA, 1990; ISBN 0-19-855168-1. [Google Scholar]
- Gillespie, R.J.; Popelier, P.L.A. Chemical Bonding and Molecular Geometry; Oxford University: New York, NY, USA, 2001; ISBN 0-19-510496-X. [Google Scholar]
- Keith, T.A. Atomic Response Properties. In The Quantum Theory of Atoms in Molecules, From Solid State to DNA and Drug Design; Wiley-VCH: Weinheim, Germany, 2007; ISBN 978-3-527-30748-7. [Google Scholar]
- Joblin, C.; Boissel, P.; Leger, A.; D’Hendecourt, L.; Defourneau, D. Infrared spectroscopy of gas-phase PAH molecules. I. Role of the physical environment. Astron. Astrophys. 1994, 281, 926–936. [Google Scholar]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadjadi, S.; Parker, Q.A. The Astrochemistry Implications of Quantum Chemical Normal Modes Vibrational Analysis. Galaxies 2018, 6, 123. https://doi.org/10.3390/galaxies6040123
Sadjadi S, Parker QA. The Astrochemistry Implications of Quantum Chemical Normal Modes Vibrational Analysis. Galaxies. 2018; 6(4):123. https://doi.org/10.3390/galaxies6040123
Chicago/Turabian StyleSadjadi, SeyedAbdolreza, and Quentin Andrew Parker. 2018. "The Astrochemistry Implications of Quantum Chemical Normal Modes Vibrational Analysis" Galaxies 6, no. 4: 123. https://doi.org/10.3390/galaxies6040123
APA StyleSadjadi, S., & Parker, Q. A. (2018). The Astrochemistry Implications of Quantum Chemical Normal Modes Vibrational Analysis. Galaxies, 6(4), 123. https://doi.org/10.3390/galaxies6040123