
Molecular Pathology and Personalized Medicine: The Dawn of a New Era in Companion Diagnostics—Practical Considerations about Companion Diagnostics for Non-Small-Cell-Lung-Cancer


 and
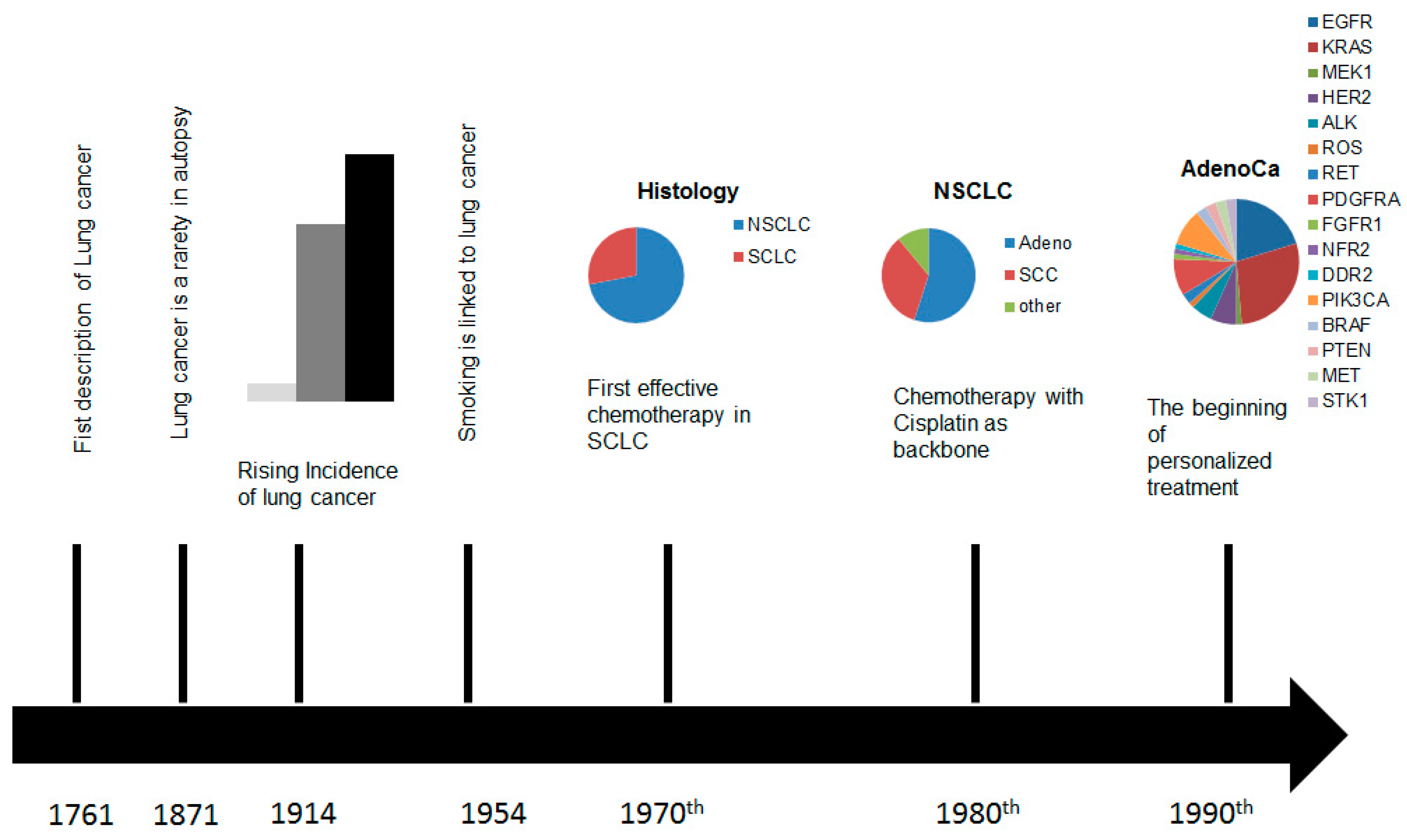
and 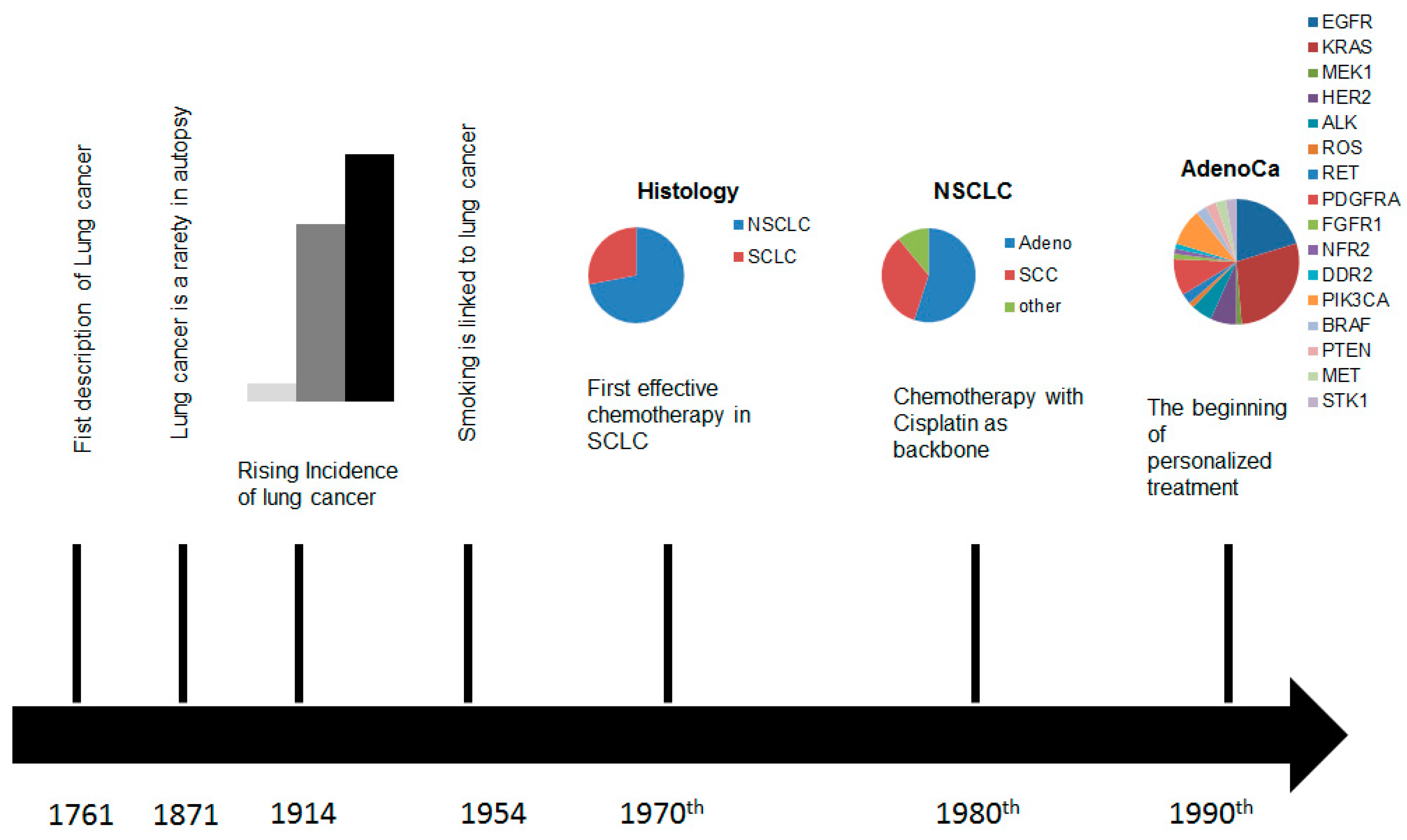{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Markers of Current Diagnostic and Therapeutic Relevance
2.1. Epidermal Growth Factor Receptor
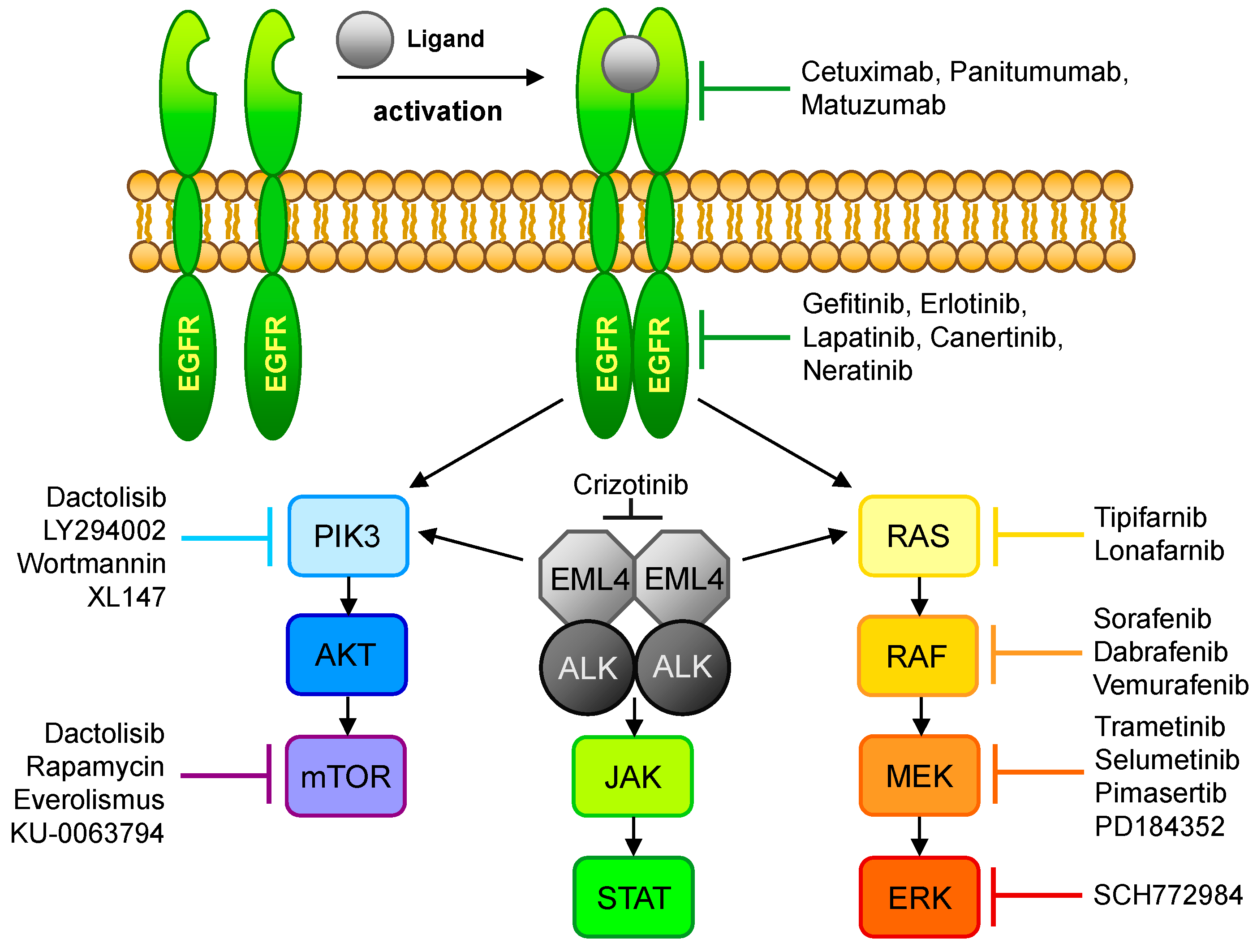
2.2. Anaplastic Lymphoma Kinase
2.3. C-Ros Oncogene 1
2.4. PD-L1
2.5. Other Biomarkers and Future Aspects
2.6. Cost Effectiveness, Facilitation, and Failures of CDx and Multiplexing
3. Summary and Conclusions
Author Contributions
Conflicts of Interest
References
- Xu, M.; Xie, Y.; Ni, S.; Liu, H. The latest therapeutic strategies after resistance to first generation epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIS) in patients with non-small cell lung cancer (NSCLC). Ann. Transl. Med. 2015, 3, 96. [Google Scholar] [PubMed]
- Thomas, A.; Liu, S.V.; Subramaniam, D.S.; Giaccone, G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat. Rev. Clin. Oncol. 2015, 12, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Kumarakulasinghe, N.B.; van Zanwijk, N.; Soo, R.A. Molecular targeted therapy in the treatment of advanced stage non-small cell lung cancer (NSCLC). Respirology 2015, 20, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Zer, A.; Leighl, N. Promising targets and current clinical trials in metastatic non-squamous NSCLC. Front. Oncol. 2014, 4, 329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lange, A.; Prenzler, A.; Frank, M.; Golpon, H.; Welte, T.; von der Schulenburg, J.M. A systematic review of the cost-effectiveness of targeted therapies for metastatic non-small cell lung cancer (NSCLC). BMC Pulm. Med. 2014, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Socinski, M.A.; Burns, T.F. Personalized treatment of EGFR mutant and alk-positive patients in NSCLC. Expert Opin. Pharmacother. 2014, 15, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Overholt, R.H.; Schmidt, I.C. Survival in primary carcinoma of the lung. N. Engl. J. Med. 1949, 240, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Ruckdeschel, J.C.; Finkelstein, D.M.; Ettinger, D.S.; Creech, R.H.; Mason, B.A.; Joss, R.A.; Vogl, S. A randomized trial of the four most active regimens for metastatic non-small-cell lung cancer. J. Clin. Oncol. 1986, 4, 14–22. [Google Scholar] [PubMed]
- Lu, S.; Yu, Y.; Chen, Z.; Ye, X.; Li, Z.; Niu, X. Maintenance therapy improves survival outcomes in patients with advanced non-small cell lung cancer: A meta-analysis of 14 studies. Lung 2015, 193, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (optimal, ctong-0802). Ann. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Sala Gonzalez, M.A. Prolonged survival with erlotinib followed by afatinib in a caucasian smoker with metastatic, poorly differentiated large cell carcinoma of the lung: A case report. Cancer Biol. Ther. 2015, 16, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Saji, H.; Kakihana, M.; Kajiwara, N.; Ohira, T.; Ikeda, N. Survival outcomes for oligometastasis in resected non-small cell lung cancer. Asian Cardiovasc. Thorac. Ann. 2015, 23, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Crvenkova, S.; Pesevska, M. Important prognostic factors for the long-term survival in non-small cell lung cancer patients treated with combination of chemotherapy and conformal radiotherapy. J. BUON 2015, 20, 775–781. [Google Scholar] [PubMed]
- Warth, A.; Muley, T.; Meister, M.; Stenzinger, A.; Thomas, M.; Schirmacher, P.; Schnabel, P.A.; Budczies, J.; Hoffmann, H.; Weichert, W. The novel histologic international association for the study of lung cancer/american thoracic society/european respiratory society classification system of lung adenocarcinoma is a stage-independent predictor of survival. J. Clin. Oncol. 2012, 30, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Olsen, D.; Jorgensen, J.T. Companion diagnostics for targeted cancer drugs—Clinical and regulatory aspects. Front. Oncol. 2014, 4, 105. [Google Scholar] [CrossRef] [PubMed]
- Marton, M.J.; Weiner, R. Practical guidance for implementing predictive biomarkers into early phase clinical studies. Biomed. Res. Int. 2013, 2013, 891391. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M.; Pfaff, O. Similarities and differences in the oncology drug approval process between fda and european union with emphasis on in vitro companion diagnostics. Clin. Cancer Res. 2014, 20, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, V.; Lochi, V.; Lüsebrink, J.; Brockmann, M.; Schildgen, O. Epidemiology of EML4-ALK translocations in a small, german non-small-cell lung cancer patient cohort. Per. Med. 2012, 9, 801–803. [Google Scholar] [CrossRef]
- Schildgen, V.; Lusebrink, J.; Appel, J.D.; Wubben, C.; Engel-Riedel, W.; Ludwig, C.; Stoelben, E.; Schildgen, O.; Brockmann, M. Identification of uncommon PIK3CA mutations in lung cancer by using pyrosequencing. Diagn. Mol. Pathol. 2013, 22, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, V.; Schulz, C.; Lüsebrink, J.; Tillmann, R.-L.; Engel-Riedel, W.; Stoelben, E.; Schildgen, O.; Brockmann, M. Combination of pyrosequencing® and sanger sequencing reveals alleged novel mutation in exon 18 of EGFR. Per. Med. 2013, 10, 201–209. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Cappuzzo, F. Predictive value of EGFR and her2 overexpression in advanced non-small-cell lung cancer. Oncogene 2009, 28 (Suppl. S1), S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chiu, H.H.; Yen, L.C.; Chang, H.J.; Chang, M.S.; Tsai, J.R.; Chen, Y.F.; Lin, S.R. Overexpression of EGFR pathway-related genes in the circulation is highly correlated with EGFR mutations and overexpression in paired cancer tissue from patients with non-small cell lung cancer. Oncol. Rep. 2010, 23, 639–645. [Google Scholar] [PubMed]
- Kawai, H.; Ishii, A.; Washiya, K.; Konno, T.; Kon, H.; Yamaya, C.; Ono, I.; Ogawa, J. Combined overexpression of EGFR and estrogen receptor alpha correlates with a poor outcome in lung cancer. Anticancer Res. 2005, 25, 4693–4698. [Google Scholar] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. Egf receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.W.; Lynch, T.J.; Haserlat, S.M.; Harris, P.L.; Okimoto, R.A.; Brannigan, B.W.; Sgroi, D.C.; Muir, B.; Riemenschneider, M.J.; Iacona, R.B.; et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: Molecular analysis of the ideal/intact gefitinib trials. J. Clin. Oncol. 2005, 23, 8081–8092. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Janne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Billah, S.; Stewart, J.; Staerkel, G.; Chen, S.; Gong, Y.; Guo, M. EGFR and KRAS mutations in lung carcinoma: Molecular testing by using cytology specimens. Cancer Cytopathol. 2011, 119, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.W.; Brannigan, B.W.; Matsuo, K.; Finkelstein, D.M.; Sordella, R.; Settleman, J.; Mitsudomi, T.; Haber, D.A. Increased prevalence of EGFR-mutant lung cancer in women and in east asian populations: Analysis of estrogen-related polymorphisms. Clin. Cancer Res. 2008, 14, 4079–4084. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Pietanza, M.C.; Johnson, M.L.; Riely, G.J.; Miller, V.A.; Sima, C.S.; Zakowski, M.F.; Rusch, V.W.; Ladanyi, M.; Kris, M.G. Incidence of EGFR exon 19 deletions and l858r in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J. Clin. Oncol. 2011, 29, 2066–2070. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Brown, C.; Gralla, R.J.; Hirsh, V.; Thongprasert, S.; Tsai, C.M.; Tan, E.H.; Ho, J.C.; Chu da, T.; Zaatar, A.; et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: A meta-analysis. J. Natl. Cancer Inst. 2013, 105, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Popat, S.; Reinmuth, N.; De Ruysscher, D.; Kerr, K.M.; Peters, S. Metastatic non-small-cell lung cancer (NSCLC): Esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. S3), 27–39. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. S1), S24–S31. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.W.; Gore, I.; Okimoto, R.A.; Godin-Heymann, N.; Sordella, R.; Mulloy, R.; Sharma, S.V.; Brannigan, B.W.; Mohapatra, G.; Settleman, J.; et al. Inherited susceptibility to lung cancer may be associated with the t790m drug resistance mutation in EGFR. Nat. Genet. 2005, 37, 1315–1316. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. Resistance to targeted therapies: Refining anticancer therapy in the era of molecular oncology. Clin. Cancer Res. 2009, 15, 7471–7478. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Ladanyi, M. KRAS mutations: An old oncogene becomes a new predictive biomarker. J. Mol. Diagn. 2008, 10, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Riely, G.J.; Nafa, K.; Ladanyi, M. KRAS mutational testing in the selection of patients for EGFR-targeted therapies. Semin. Diagn. Pathol. 2008, 25, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Kris, M.G.; Rosenbaum, D.; Marks, J.; Li, A.; Chitale, D.A.; Nafa, K.; Riedel, E.R.; Hsu, M.; Pao, W.; et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 5731–5734. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, J.; Engel-Riedel, W.; Prickartz, A.; Ludwig, C.; Schildgen, O.; Tillmann, R.L.; Stoelben, E.; Brockmann, M.; Schildgen, V. Combined point mutation in KRAS or EGFR genes and EML4-ALK translocation in lung cancer patients. Future Oncol. 2014, 10, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Wang, F.; Shao, Q.; Zhang, X.; Duan, L.P.; Zhang, L.; Shao, J.Y. Detection of EML4-ALK fusion gene in chinese non-small cell lung cancer by using a sensitive quantitative real-time reverse transcriptase PCR technique. Diagn. Mol. Pathol. 2014, 23, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Robesova, B.; Bajerova, M.; Liskova, K.; Skrickova, J.; Tomiskova, M.; Pospisilova, S.; Mayer, J.; Dvorakova, D. Taqman based real time pcr assay targeting EML4-ALK fusion transcripts in NSCLC. Lung Cancer 2014, 85, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Choi, Y.L.; Soda, M.; Inamura, K.; Togashi, Y.; Hatano, S.; Enomoto, M.; Takada, S.; Yamashita, Y.; Satoh, Y.; et al. Multiplex reverse transcription-pcr screening for EML4-ALK fusion transcripts. Clin. Cancer Res. 2008, 14, 6618–6624. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Karachaliou, N.; Peg, V.; Gimenez-Capitan, A.; Rosell, R. Concordance of IHC, fish and RT-PCR for EML4-ALK rearrangements. Transl. Lung Cancer Res. 2014, 3, 70–74. [Google Scholar] [PubMed]
- Tuononen, K.; Maki-Nevala, S.; Sarhadi, V.K.; Wirtanen, A.; Ronty, M.; Salmenkivi, K.; Andrews, J.M.; Telaranta-Keerie, A.I.; Hannula, S.; Lagstrom, S.; et al. Comparison of targeted next-generation sequencing (NGS) and real-time pcr in the detection of EGFR, KRAS, and braf mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Genes Chromosomes Cancer 2013, 52, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Armengol, G.; Sarhadi, V.K.; Ronty, M.; Tikkanen, M.; Knuuttila, A.; Knuutila, S. Driver gene mutations of non-small-cell lung cancer are rare in primary carcinoids of the lung: Ngs study by ion torrent. Lung 2015, 193, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Morris-Rosendahl, D.J.; Kaindl, A.M. What next-generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH). Mol. Cell. Probes 2015, 29, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ardeshirdavani, A.; Souche, E.; Dehaspe, L.; Van Houdt, J.; Vermeesch, J.R.; Moreau, Y. Ngs-logistics: Federated analysis of NGS sequence variants across multiple locations. Genome Med. 2014, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Ye, K.; Gu, Z.; Sun, X. Diminishing returns in next-generation sequencing (NGS) transcriptome data. Gene 2015, 557, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Jiang, L.; Chong, Z.; Gong, Q.; Li, H.; Li, C.; Tao, Y.; Zheng, C.; Zhai, W.; Turissini, D.; et al. Pseudo-sanger sequencing: Massively parallel production of long and near error-free reads using NGS technology. BMC Genom. 2013, 14, 711. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.R.; Kerick, M.; Timmermann, B.; Isau, M. The power of NGS technologies to delineate the genome organization in cancer: From mutations to structural variations and epigenetic alterations. Cancer Metast. Rev. 2011, 30, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, J.L.; Niemela, J.E.; Fleisher, T.A.; Rosenzweig, S.D. Targeted NGS: A cost-effective approach to molecular diagnosis of pids. Front. Immunol. 2014, 5, 531. [Google Scholar] [CrossRef] [PubMed]
- Fenizia, F.; De Luca, A.; Pasquale, R.; Sacco, A.; Forgione, L.; Lambiase, M.; Iannaccone, A.; Chicchinelli, N.; Franco, R.; Rossi, A.; et al. EGFR mutations in lung cancer: From tissue testing to liquid biopsy. Future Oncol. 2015, 11, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Von Laffert, M.; Penzel, R.; Schirmacher, P.; Warth, A.; Lenze, D.; Hummel, M.; Dietel, M. Multicenter ALK testing in non-small-cell lung cancer: Results of a round robin test. J. Thorac. Oncol. 2014, 9, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Maus, M.K.; Desai, S.J.; Beckett, L.A.; Stephens, C.; Huang, E.; Hsiang, J.; Zeger, G.; Danenberg, K.D.; Astrow, S.H.; et al. Large-scale screening and molecular characterization of EML4-ALK fusion variants in archival non-small-cell lung cancer tumor specimens using quantitative reverse transcription polymerase chain reaction assays. J. Thorac. Oncol. 2014, 9, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.K.; Stephens, C.; Zeger, G.; Grimminger, P.P.; Huang, E. Identification of novel variant of EML4-ALK fusion gene in NSCLC: Potential benefits of the RT-PCR method. Int. J. Biomed. Sci. 2012, 8, 1–6. [Google Scholar] [PubMed]
- To, K.F.; Tong, J.H.; Yeung, K.S.; Lung, R.W.; Law, P.P.; Chau, S.L.; Kang, W.; Tong, C.Y.; Chow, C.; Chan, A.W.; et al. Detection of alk rearrangement by immunohistochemistry in lung adenocarcinoma and the identification of a novel EML4-ALK variant. J. Thorac. Oncol. 2013, 8, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Penzel, R.; Schirmacher, P.; Warth, A. A novel EML4-ALK variant: Exon 6 of EML4 fused to exon 19 of ALK. J. Thorac. Oncol. 2012, 7, 1198–1199. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, N.; Bandoh, S.; Ishii, T.; Tadokoro, A.; Watanabe, N.; Takahama, T.; Haba, R.; Imataki, O.; Dobashi, H.; Matsunaga, T. Detection of EML4-ALK fusion genes in a few cancer cells from transbronchial cytological specimens utilizing immediate cytology during bronchoscopy. Lung Cancer 2012, 77, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Balke-Want, H.; Malchers, F.; Peifer, M.; Sos, M.L.; Koker, M.; Meder, L.; Lovly, C.M.; Heukamp, L.C.; Pao, W.; et al. Differential protein stability and alk inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. 2012, 18, 4682–4690. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Mino-Kenudson, M.; Dacic, S.; Yeap, B.Y.; Shaw, A.; Barletta, J.A.; Stubbs, H.; Law, K.; Lindeman, N.; Mark, E.; et al. Unique clinicopathologic features characterize alk-rearranged lung adenocarcinoma in the western population. Clin. Cancer Res. 2009, 15, 5216–5223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, S.; Yang, X.; Yang, J.; Zhou, Q.; Yin, L.; An, S.; Lin, J.; Chen, S.; Xie, Z.; et al. Fusion of EML4 and alk is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with alk expression. Mol. Cancer 2010, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Robesova, B.; Bajerova, M.; Hausnerova, J.; Skrickova, J.; Tomiskova, M.; Dvorakova, D. Identification of atypical atrnl1 insertion to EML4-ALK fusion gene in NSCLC. Lung Cancer 2015, 87, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Streubel, A.; Grah, C.; Stephan-Falkenau, S.; Mairinger, T.; Wagner, F. A rare case of an EML4-ALK-rearranged lung adenocarcinoma missed by in situ-hybridization but detected by RT-PCR. J. Clin. Pathol. 2014, 67, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Takenoyama, M.; Taguchi, K.; Toyozawa, R.; Inamasu, E.; Kojo, M.; Shiraishi, Y.; Morodomi, Y.; Takenaka, T.; Hirai, F.; et al. An extremely rare case of small-cell lung cancer harboring variant 2 of the EML4-ALK fusion gene. Lung Cancer 2013, 81, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Batist, G.; Wu, J.H.; Spatz, A.; Miller, W.H., Jr.; Cocolakis, E.; Rousseau, C.; Diaz, Z.; Ferrario, C.; Basik, M. Resistance to cancer treatment: The role of somatic genetic events and the challenges for targeted therapies. Front. Pharmacol. 2011, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. Met amplification occurs with or without t790m mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.B.; Nguyen, K.S.; Cho, B.C.; Sequist, L.V.; Jackman, D.M.; Riely, G.J.; Yeap, B.Y.; Halmos, B.; Kim, J.H.; Janne, P.A.; et al. Effects of erlotinib in EGFR mutated non-small cell lung cancers with resistance to gefitinib. Clin. Cancer Res. 2008, 14, 7060–7067. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed]
- Lozano, M.D.; Labiano, T.; Zudaire, I.; Subtil, J.C.; Gurpide, A.; Echeveste, J.I.; Zulueta, J.J.; Martin-Algarra, S.; Perez-Gracia, J.L. Variations in molecular profile in NSCLC can be analyzed using cytological samples: Development of EGFR resistance mutations and coexistence of alk-eml4 translocation in an EGFR-sensitive patient. Int. J. Surg. Pathol. 2015, 23, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel alk secondary mutation and EGFR signaling cause resistance to alk kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [PubMed]
- Abbott Molecular. Available online: https://www.abbottmolecular.com/us/products/vysis-lsi-alk-dual-color-break-apart-rearrangement-probe.html (accessed on 26 November 2015).
- Borrelli, N.; Giannini, R.; Proietti, A.; Ali, G.; Pelliccioni, S.; Niccoli, C.; Lucchi, M.; Melfi, F.; Mussi, A.; Basolo, F.; et al. KIF5B/RET fusion gene analysis in a selected series of cytological specimens of EGFR, KRAS and EML4-ALK wild-type adenocarcinomas of the lung. Lung Cancer 2013, 81, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Calio, A.; Nottegar, A.; Gilioli, E.; Bria, E.; Pilotto, S.; Peretti, U.; Kinspergher, S.; Simionato, F.; Pedron, S.; Knuutila, S.; et al. Alk/eml4 fusion gene may be found in pure squamous carcinoma of the lung. J. Thorac. Oncol. 2014, 9, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, X.; Liu, J.; Zha, J.; Pei, L. Evaluation of EML4-ALK fusion proteins in non-small cell lung cancer using small molecule inhibitors. Neoplasia 2011, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mano, H. Non-solid oncogenes in solid tumors: Eml4-alk fusion genes in lung cancer. Cancer Sci. 2008, 99, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Mano, H.; Takeuchi, K. EML4-ALK fusion in lung. Am. J. Pathol. 2010, 176, 1552–1553. [Google Scholar] [CrossRef] [PubMed]
- Von Laffert, M.; Warth, A.; Penzel, R.; Schirmacher, P.; Jonigk, D.; Kreipe, H.; Schildhaus, H.U.; Merkelbach-Bruse, S.; Buttner, R.; Reu, S.; et al. Anaplastic lymphoma kinase (ALK) gene rearrangement in non-small cell lung cancer (NSCLC): Results of a multi-centre alk-testing. Lung Cancer 2013, 81, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Jurmeister, P.; Lenze, D.; Berg, E.; Mende, S.; Schaper, F.; Kellner, U.; Herbst, H.; Sers, C.; Budczies, J.; Dietel, M.; et al. Parallel screening for alk, met and ros1 alterations in non-small cell lung cancer with implications for daily routine testing. Lung Cancer 2015, 87, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Pekar-Zlotin, M.; Hirsch, F.R.; Soussan-Gutman, L.; Ilouze, M.; Dvir, A.; Boyle, T.; Wynes, M.; Miller, V.A.; Lipson, D.; Palmer, G.A.; et al. Fluorescence in situ hybridization, immunohistochemistry, and next-generation sequencing for detection of EML4-ALK rearrangement in lung cancer. Oncologist 2015, 20, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Rico, G.; Aviles-Salas, A.; Segura-Gonzalez, M.; Espinosa-Garcia, A.M.; Ramirez-Tirado, L.A.; Morales-Oyarvide, V.; Rojas-Marin, C.; Cardona, A.F.; Arrieta, O. Diagnosis of EML4-ALK translocation with fish, immunohistochemistry, and real-time polymerase chain reaction in patients with non-small cell lung cancer. Am. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Just, P.A.; Cazes, A.; Audebourg, A.; Cessot, A.; Pallier, K.; Danel, C.; Vacher-Lavenu, M.C.; Laurent-Puig, P.; Terris, B.; Blons, H. Histologic subtypes, immunohistochemistry, fish or molecular screening for the accurate diagnosis of alk-rearrangement in lung cancer: A comprehensive study of caucasian non-smokers. Lung Cancer 2012, 76, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Wang, L.; Hasanovic, A.; Suehara, Y.; Lipson, D.; Stephens, P.; Ross, J.; Miller, V.; Ginsberg, M.; Zakowski, M.F.; et al. Response to cabozantinib in patients with ret fusion-positive lung adenocarcinomas. Cancer Discov. 2013, 3, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, J.T. Companion diagnostic assays for pd-1/pd-l1 checkpoint inhibitors in NSCLC. Expert Rev. Mol. Diagn. 2015, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Zimmer, R. Count ratio model reveals bias affecting NGS fold changes. Nucleic Acids Res. 2015, 43, e136. [Google Scholar] [CrossRef] [PubMed]
- Korfhage, C.; Fricke, E.; Meier, A. Parallel WGA and WTA for comparative genome and transcriptome NGS analysis using tiny cell numbers. Curr. Protoc. Mol. Biol. 2015, 111, 11–18. [Google Scholar]
- De Brevern, A.G.; Meyniel, J.P.; Fairhead, C.; Neuveglise, C.; Malpertuy, A. Trends in it innovation to build a next generation bioinformatics solution to manage and analyse biological big data produced by NGS technologies. Biomed. Res. Int. 2015, 2015, 904541. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Kantar, M. Harnessing NGS and big data optimally: Comparison of mirna prediction from assembled versus non-assembled sequencing data-the case of the grass aegilops tauschii complex genome. OMICS 2015, 19, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Dacheva, D.; Dodova, R.; Popov, I.; Goranova, T.; Mitkova, A.; Mitev, V.; Kaneva, R. Validation of an NGS approach for diagnostic BRCA1/BRCA2 mutation testing. Mol. Diagn. Ther. 2015, 19, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Glotov, A.S.; Kazakov, S.V.; Zhukova, E.A.; Alexandrov, A.V.; Glotov, O.S.; Pakin, V.S.; Danilova, M.M.; Poliakova, I.V.; Niyazova, S.S.; Chakova, N.N.; et al. Targeted next-generation sequencing (NGS) of nine candidate genes with custom ampliseq in patients and a cardiomyopathy risk group. Clin. Chim. Acta 2015, 446, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.W. Thoughts on NGS, alternative splicing and what we still need to know. RNA 2015, 21, 683–684. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boland, P.M.; Ruth, K.; Matro, J.M.; Rainey, K.L.; Fang, C.Y.; Wong, Y.N.; Daly, M.B.; Hall, M.J. Genetic counselors’ (GC) knowledge, awareness, understanding of clinical next-generation sequencing (NGS) genomic testing. Clin. Genet. 2015, 88, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S. Ensuring confident detection of disease-linked variants with NGS. MLO Med. Lab. Obs. 2014, 46, 26. [Google Scholar]
- Lighten, J.; van Oosterhout, C.; Bentzen, P. Critical review of NGS analyses for de novo genotyping multigene families. Mol Ecol 2014, 23, 3957–3972. [Google Scholar] [CrossRef] [PubMed]
- Elbaidouri, M.; Chaparro, C.; Panaud, O. Use of next generation sequencing (NGS) technologies for the genome-wide detection of transposition. Methods Mol. Biol. 2013, 1057, 265–274. [Google Scholar] [PubMed]
- Xia, J.; Wang, Q.; Jia, P.; Wang, B.; Pao, W.; Zhao, Z. Ngs catalog: A database of next generation sequencing studies in humans. Hum. Mutat. 2012, 33, E2341–E2355. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Arantes, A.S.; Liao, X.; Stothard, P. In-depth annotation of snps arising from resequencing projects using NGS-snp. Bioinformatics 2011, 27, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Califano, R.; Morgillo, F.; de Mello, R.A.; Mountzios, G. Role of mesenchymal-epithelial transition amplification in resistance to anti-epidermal growth factor receptor agents. Ann. Transl. Med. 2015, 3, 81. [Google Scholar] [PubMed]
- Schildgen, V.; Schildgen, O. How is a molecular polymorphism defined? Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plönes, T.; Engel-Riedel, W.; Stoelben, E.; Limmroth, C.; Schildgen, O.; Schildgen, V. Molecular Pathology and Personalized Medicine: The Dawn of a New Era in Companion Diagnostics—Practical Considerations about Companion Diagnostics for Non-Small-Cell-Lung-Cancer. J. Pers. Med. 2016, 6, 3. https://doi.org/10.3390/jpm6010003
Plönes T, Engel-Riedel W, Stoelben E, Limmroth C, Schildgen O, Schildgen V. Molecular Pathology and Personalized Medicine: The Dawn of a New Era in Companion Diagnostics—Practical Considerations about Companion Diagnostics for Non-Small-Cell-Lung-Cancer. Journal of Personalized Medicine. 2016; 6(1):3. https://doi.org/10.3390/jpm6010003
Chicago/Turabian StylePlönes, Till, Walburga Engel-Riedel, Erich Stoelben, Christina Limmroth, Oliver Schildgen, and Verena Schildgen. 2016. "Molecular Pathology and Personalized Medicine: The Dawn of a New Era in Companion Diagnostics—Practical Considerations about Companion Diagnostics for Non-Small-Cell-Lung-Cancer" Journal of Personalized Medicine 6, no. 1: 3. https://doi.org/10.3390/jpm6010003
APA StylePlönes, T., Engel-Riedel, W., Stoelben, E., Limmroth, C., Schildgen, O., & Schildgen, V. (2016). Molecular Pathology and Personalized Medicine: The Dawn of a New Era in Companion Diagnostics—Practical Considerations about Companion Diagnostics for Non-Small-Cell-Lung-Cancer. Journal of Personalized Medicine, 6(1), 3. https://doi.org/10.3390/jpm6010003