
Effectiveness and Clinical Outcomes of PGT-M Using Karyomapping for Successful Pregnancy and Birth in Various Types of Charcot–Marie–Tooth Disease

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Characteristics
2.2. Preclinical Test for Karyomapping
2.3. Embryo Biopsy and Whole-Genome Amplification
2.4. Karyomapping for PGT-M
2.5. Embryo Transfer and In Vitro Fertilization Outcome
2.6. Prenatal Diagnosis
3. Results
3.1. Preclinical Test Using DNA of the Affected Family
3.2. Clinical PGT for CMT
3.2.1. CMT1A
3.2.2. CMT1B
3.2.3. CMT2A
3.2.4. CMT2
3.2.5. CMTX1
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CMT | Charcot–Marie–Tooth disease |
| PGT-M | Preimplantation genetic testing for monogenic disorders |
| SNPs | Single-nucleotide polymorphisms |
| EDTA | Ethylenediaminetetraacetic acid |
| IVF | In vitro fertilization |
| TE | Trophectoderm |
| WGA | Whole-genome amplification |
| PBS | Phosphate-buffered saline |
| CMA | Chromosomal microarray analysis |
References
- Beloribi-Djefaflia, S.; Attarian, S. Treatment of Charcot–Marie–Tooth neuropathies. Rev. Neurol. 2023, 179, 35–48. [Google Scholar] [CrossRef]
- Kim, H.J.; Nam, S.H.; Kwon, H.M.; Lim, S.O.; Park, J.H.; Kim, H.S.; Kim, S.B.; Lee, K.S.; Lee, J.E.; Choi, B.O.; et al. Genetic and clinical spectrums in Korean Charcot–Marie–Tooth disease patients with myelin protein zero mutations. Mol. Genet. Genomic Med. 2021, 9, e1678. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Kuo, Y.; Guan, S.; Wang, N.; Lian, Y.; Huang, J.; Zhi, X.; Liu, P.; Li, R.; et al. Clinical practice and outcomes of preimplantation genetic testing for CMT1A using a novel direct detection method. Heliyon 2023, 9, e22196. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.M.; Choi, B.O. Analyzing clinical and genetic aspects of axonal Charcot–Marie–Tooth disease. J. Genet. Med. 2021, 18, 83–93. [Google Scholar] [CrossRef]
- Okamoto, Y.; Takashima, H. The current state of Charcot–Marie–Tooth disease treatment. Genes 2023, 14, 1391. [Google Scholar] [CrossRef]
- Cortese, A.; Wilcox, J.E.; Polke, J.M.; Poh, R.; Skorupinska, M.; Rossor, A.M.; Laura, M.; Tomaselli, P.J.; Houlden, H.; Shy, M.E.; et al. Targeted next-generation sequencing panels in the diagnosis of Charcot–Marie–Tooth disease. Neurology 2020, 94, e51–e61. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.W.; Liu, Y.L.; Chen, C.H. Targeting myotonic dystrophy by preimplantation genetic diagnosis-karyomapping. Taiwan. J. Obstet. Gynecol. 2019, 58, 891–894. [Google Scholar] [CrossRef]
- Renwick, P.; Ogilvie, C.M. Preimplantation genetic diagnosis for monogenic diseases: Overview and emerging issues. Expert Rev. Mol. Diagn. 2007, 7, 33–43. [Google Scholar] [CrossRef]
- Borgulová, I.; Putzová, M.; Soldatova, I.; Stejskal, D. Preimplantation genetic diagnosis of X-linked Charcot–Marie–Tooth disease by indirect linkage analysis. Med. Clin. 2018, 150, 215–219. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, M.J.; Ko, D.S.; Jeon, E.J.; Kim, J.Y.; Kang, I.S. Preimplantation genetic diagnosis for Charcot–Marie–Tooth disease. Clin. Exp. Reprod. Med. 2013, 40, 163–168. [Google Scholar] [CrossRef]
- Handyside, A.H.; Harton, G.L.; Mariani, B.; Thornhill, A.R.; Affara, N.; Shaw, M.A.; Griffin, D.K. Karyomapping: A universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med. Genet. 2010, 47, 651–658. [Google Scholar] [CrossRef]
- Thornhill, A.R.; Handyside, A.H.; Ottolini, C.; Natesan, S.A.; Taylor, J.; Sage, K.; Harton, G.; Cliffe, K.; Affara, N.; Konstantinidis, M.; et al. Karyomapping—A comprehensive means of simultaneous monogenic and cytogenetic PGD: Comparison with standard approaches in real time for Marfan syndrome. J. Assist. Reprod. Genet. 2015, 32, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, M.; Prates, R.; Goodall, N.N.; Fischer, J.; Tecson, V.; Lemma, T.; Chu, B.; Jordan, A.; Armenti, E.; Wells, D.; et al. Live births following Karyomapping of human blastocysts: Experience from clinical application of the method. Reprod. Biomed. Online 2015, 31, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Park, S.O.; Hong, Y.S.; Park, E.A.; Lee, Y.B.; Choi, B.O.; Lee, K.A.; Yu, E.J.; Kang, I.S. Clinical application of genome-wide single nucleotide polymorphism genotyping and karyomapping for preimplantation genetic testing of Charcot–Marie–Tooth disease. J. Genet. Med. 2022, 19, 7–13. [Google Scholar] [CrossRef]
- ASRM Practice Committee. Indications and management of preimplantation genetic testing for monogenic conditions: A committee opinion. Fertil. Steril. 2023, 120, 61–71. [Google Scholar] [CrossRef]
- Gardner, D.K.; Lane, M.; Stevens, J.; Schlenker, T.; Schoolcraft, W.B. Blastocyst score affects implantation and pregnancy outcome: Towards a single blastocyst transfer. Fertil. Steril. 2000, 73, 1155–1158. [Google Scholar] [CrossRef]
- ACOG Committee on Genetics. Prenatal diagnostic testing for genetic disorders. Obstet. Gynecol. 2016, 127, 978–981. [Google Scholar] [CrossRef]
- Ott, J. Analysis of Human Genetic Linkage, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 1999. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Natesan, S.A.; Bladon, A.J.; Coskun, S.; Qubbaj, W.; Prates, R.; Munne, S.; Coonen, E.; Dreesen, J.C.F.M.; Stevens, S.J.C.; Paulussen, A.D.C.; et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet. Med. 2014, 16, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, L.; Shi, H.; Niu, W.; Wang, Y.; Bu, B.; Liu, Y.; Bao, X.; Song, W.; Jin, H.; et al. Preimplantation genetic testing for four families with severe combined immunodeficiency: Three unaffected livebirths. Orphanet J. Rare Dis. 2025, 20, 14. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Xu, J.; Niu, W.; Liu, Y.; Shi, H.; Yao, G.; Shi, S.; Li, G.; Song, W.; Jin, H.; et al. Live births following preimplantation genetic testing for dynamic mutation diseases by karyomapping: A report of three cases. J. Assist. Reprod. Genet. 2020, 37, 539–548. [Google Scholar] [CrossRef]
- Carvalho, F.; Moutou, C.; Dimitriadou, E.; Dreesen, J.; Giménez, C.; Goossens, V.; Kakourou, G.; Vermeulen, N.; Zuccarello, D.; De Rycke, M.; et al. ESHRE PGT Consortium good practice recommendations for the detection of monogenic disorders. Hum. Reprod. Open 2020, 2020, hoaa018. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, R.; Maione, A.; Vallefuoco, A.; Sorrentino, U.; Zuccarello, D. Preimplantation genetic testing for genetic diseases: Limits and review of current literature. Genes 2023, 14, 2095. [Google Scholar] [CrossRef]
- Ben-Nagi, J.; Wells, D.; Doye, K.; Loutradi, K.; Exeter, H.; Drew, E.; Alfarawati, S.; Naja, R.; Serhal, P. Karyomapping: A single centre’s experience from application of methodology to ongoing pregnancy and live-birth rates. Reprod. Biomed. Online 2017, 35, 264–271. [Google Scholar] [CrossRef]
- Kim, M.J.; Park, S.O.; Hong, Y.S.; Han, G.; Park, E.A.; Lee, K.A.; Yu, E.J.; Kang, I.S. PGT-M strategy using genome-wide haplotype karyomapping analysis for patients with de novo mutations. Fertil. Steril. 2022, 118, e253–e254. [Google Scholar] [CrossRef]
- Alteri, A.; Cermisoni, G.C.; Pozzoni, M.; Gaeta, G.; Cavoretto, P.I.; Viganò, P. Obstetric, neonatal, and child health outcomes following embryo biopsy for preimplantation genetic testing. Hum. Reprod. Update 2023, 29, 291–306. [Google Scholar] [CrossRef] [PubMed]
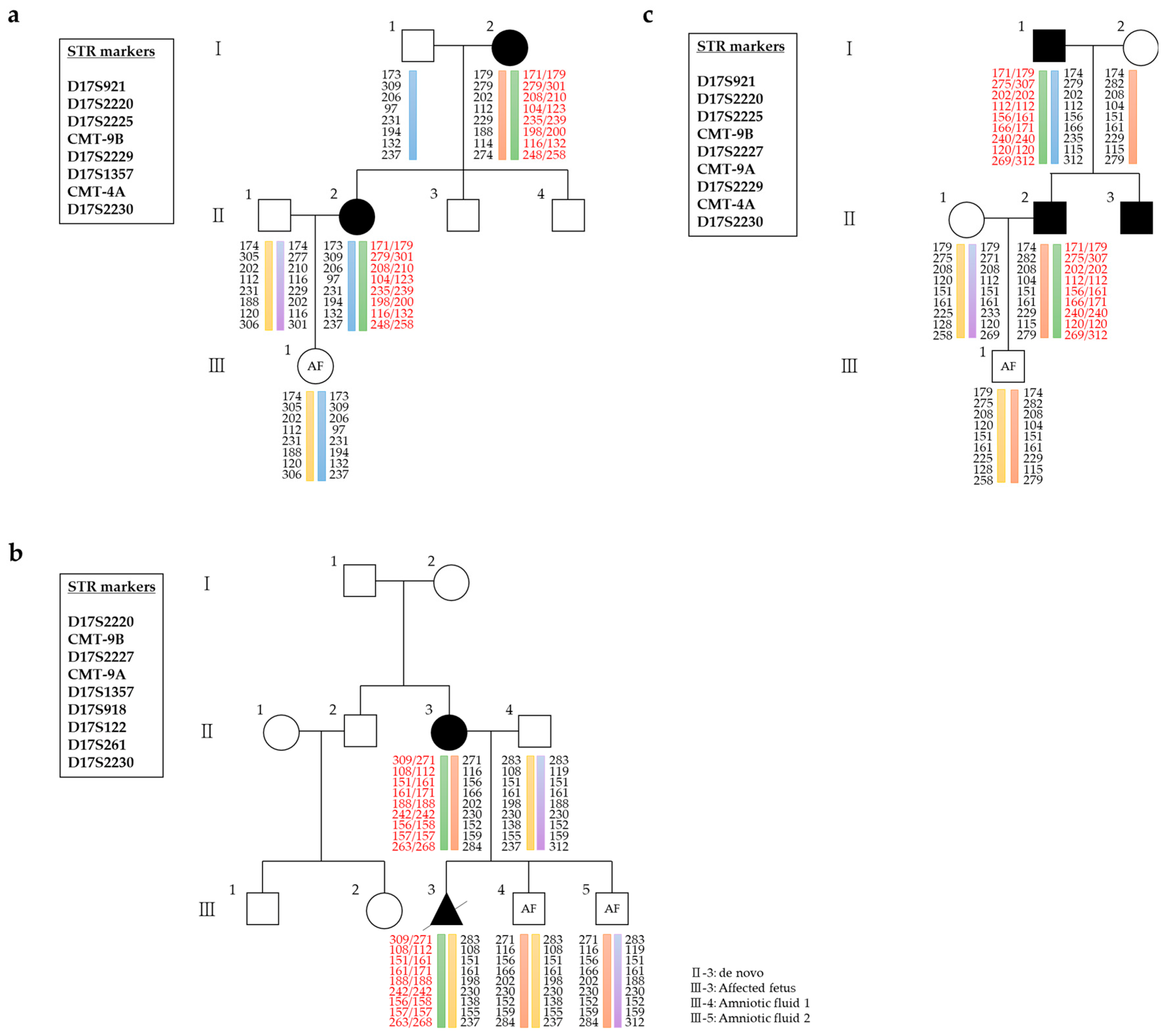


{kind=link}
{kind=link}
| Couple | Affected Partner | Female’s Age (yr) | Phenotype | Inheritance Mode | Gene | Exon | Variant | Amino Acid Change | Reference Sequence Accession Numbers | Classification |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 33 | CMT1A | AD | PMP22 | exon 1–5 | 17p11-p12 duplication | No amino acid change(duplication) | NM_000304.3 | Pathogenic |
| 2 | Male | 31 | CMT1A | AD | PMP22 | exon 1–5 | 17p11-p12 duplication | No amino acid change(duplication) | NM_000304.3 | Pathogenic |
| 3 | Male | 38 | CMT1A | AD | PMP22 | exon 1–5 | 17p11-p12 duplication | No amino acid change(duplication) | NM_000304.3 | Pathogenic |
| 4 | Female | 34 | CMT1A | AD | PMP22 | exon 1–5 | 17p11-p12 duplication | No amino acid change(duplication) | NM_000304.3 | Pathogenic |
| 5 | Female | 40 | CMT1A | AD | PMP22 | exon 1–5 | 17p11-p12 duplication | No amino acid change(duplication) | NM_000304.3 | Pathogenic |
| 6 | Male | 33 | CMT1A | AD | PMP22 | exon 1–5 | 17p11-p12 duplication | No amino acid change(duplication) | NM_000304.3 | Pathogenic |
| 7 | Female | 34 | CMT1A | AD | PMP22 | exon 1–5 | 17p11-p12 duplication | No amino acid change(duplication) | NM_000304.3 | Pathogenic |
| 8 | Male | 32 | CMT1B | AD | MPZ | exon 3 | c.410G>A | Gly137Asp | NM_000530.8 | Pathogenic |
| 9 | Male | 29 | CMT1B | AD | MPZ | exon 3 | c.242A>G | His81Arg | NM_000530.8 | Pathogenic |
| 10 | Male | 30 | CMT2A | AD | MFN2 | exon 9 | c.839G>A | Arg280His | NM_014874.4 | Pathogenic |
| 11 | Female | 31 | CMT2 | AD | MYH14 | exon 23 | c.2822G>T | Arg941Leu | NM_001077186.2 | Pathogenic |
| 12 | Female | 33 | CMTX1 | XLD | GJB1 | exon 2 | c.283G>A | Val95Met | NM_000166.6 | Pathogenic |
| 13 | Female | 36 | CMTX1 | XLD | GJB1 | exon 2 | c.457T>G | Phe153Val | NM_000166.6 | Likely pathogenic |
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | Case 11 | Case 12 | Case 13 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | CMT1A | CMT1A | CMT1A | CMT1A | CMT1A | CMT1A | CMT1A | CMT1B | CMT1B | CMT2A | CMT2 | CMTX1 | CMTX1 | |
| Affected partner | Male | Male | Male | Female | Female | Male | Female | Male | Male | Male | Female | Female | Female | |
| Reference | Affected mother | Affected father | Affected mother | Affected mother | Amniotic fluid from the affected baby | Affected father | Affected father | Affected father | Affected mother | Affected father | Affected mother | Affected father | Affected mother | |
| Target gene | PMP22 | PMP22 | PMP22 | PMP22 | PMP22 | PMP22 | PMP22 | MPZ | MPZ | MFN2 | MYH14 | GJB1 | GJB1 | |
| 5′ region | 20/351 | 25/351 | 27/351 | 21/351 | 73/351 | 28/351 | 18/351 | 11/152 | 15/152 | 44/350 | 10/96 | 43/102 | 17/102 | |
| Main region | 1/15 | 0/15 | 0/15 | 0/15 | 6/15 | 0/15 | 0/15 | 0/0 | 0/0 | 0/6 | 2/14 | 0/0 | 0/0 | |
| 3′ region | 6/233 | 4/233 | 3/233 | 3/233 | 37/233 | 25/233 | 14/233 | 24/193 | 23/193 | 7/200 | 23/153 | 5/63 | 10/63 |
| Clinical Data | Types | Total | ||||
|---|---|---|---|---|---|---|
| CMT1A | CMT1B | CMT2A | CMT2 | CMTX1 | ||
| No. of couples treated | 7 | 2 | 1 | 1 | 2 | 13 |
| Maternal age (mean, years) | 35.4 ± 2.9 | 31.0 ± 1.4 | 31 | 30 | 35.0 ± 1.4 | 33.9 ± 3.1 |
| No. of OPU cycles performed | 22 | 2 | 1 | 2 | 4 | 31 |
| No. of oocytes retrieved | 367 | 29 | 41 | 43 | 56 | 536 |
| No. of oocytes fertilized (%) | 211 (57.5) | 21 (72.4) | 30 (73.2) | 28 (65.1) | 42 (75.0) | 332 (68.6) |
| No. of embryos biopsied (%) | 90 (42.7) | 14 (66.7) | 11 (36.7) | 12 (42.9) | 23 (54.8) | 150 (45.2) |
| No. of embryos diagnosed (%) | 88 (97.8%) | 14 (100%) | 11 (100%) | 12 (100%) | 23 (100%) | 148 (98.7) |
| Normal rate by CMT (%) | 41 (45.6) | 9 (64.3) | 6 (54.5) | 6 (50.0) | 13 (56.5) | 75 (50.0) |
| Abnormal rate by CMT (%) | 47 (52.2) | 5 (35.7) | 5 (45.5) | 6 (50.0) | 10 (43.5) | 73 (48.7) |
| No result (%) | 2 (2.2) | 0 | 0 | 0 | 0 | 2 (1.3) |
| No. of embryo transfer cycles | 9 | 2 | 2 | 2 | 4 | 19 |
| No. of embryos transferred (mean) | 10 (1.1) | 2 (1.0) | 2 (1.0) | 2 (1.0) | 5 (1.2) | 21 (1.1) |
| Clinical pregnancy rate per embryo transfer (%) | 8/10 (80.0%) | 2/2 (100%) | 2/2 (100%) | 1/2 (50.0%) | 2/5 (40.0%) | 15/21 (71.4%) |
| Miscarriage rate | 0 | 0 | 0 | 0 | 0 | 0 |
| Live birth rate | 8/8 (100%) | 2/2 (100%) | 2/2 (100%) | 1/1 (100%) | 2/2 (100%) | 15/15 (100%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, G.; Kim, M.J.; Hong, Y.S.; Lee, S.; Lee, J.; Lee, Y.R.; Lee, H.-S.; Lee, K.A.; Choi, B.-O.; Yu, E.J.; et al. Effectiveness and Clinical Outcomes of PGT-M Using Karyomapping for Successful Pregnancy and Birth in Various Types of Charcot–Marie–Tooth Disease. J. Pers. Med. 2025, 15, 268. https://doi.org/10.3390/jpm15070268
Han G, Kim MJ, Hong YS, Lee S, Lee J, Lee YR, Lee H-S, Lee KA, Choi B-O, Yu EJ, et al. Effectiveness and Clinical Outcomes of PGT-M Using Karyomapping for Successful Pregnancy and Birth in Various Types of Charcot–Marie–Tooth Disease. Journal of Personalized Medicine. 2025; 15(7):268. https://doi.org/10.3390/jpm15070268
Chicago/Turabian StyleHan, Gaeul, Min Jee Kim, Ye Seul Hong, Shinhyung Lee, Jieun Lee, Ye Ryeong Lee, Hyoung-Song Lee, Kyung Ah Lee, Byung-Ok Choi, Eun Jeong Yu, and et al. 2025. "Effectiveness and Clinical Outcomes of PGT-M Using Karyomapping for Successful Pregnancy and Birth in Various Types of Charcot–Marie–Tooth Disease" Journal of Personalized Medicine 15, no. 7: 268. https://doi.org/10.3390/jpm15070268
APA StyleHan, G., Kim, M. J., Hong, Y. S., Lee, S., Lee, J., Lee, Y. R., Lee, H.-S., Lee, K. A., Choi, B.-O., Yu, E. J., & Kang, I. S. (2025). Effectiveness and Clinical Outcomes of PGT-M Using Karyomapping for Successful Pregnancy and Birth in Various Types of Charcot–Marie–Tooth Disease. Journal of Personalized Medicine, 15(7), 268. https://doi.org/10.3390/jpm15070268