
Clinical Pharmacogenetics: Results After Implementation of Preemptive Tests in Daily Routine

 , , , , , and
, , , , , and
Abstract
1. Introduction
1.1. Strategies and Methodology for PGx Implementation in Clinical Routine
1.2. Relevant Drug–Gene Interactions
1.3. Relevance of Allelic, Genotypic, and Phenotypic Frequencies in PGx Implementation in Clinical Practice
2. Materials and Methods
2.1. Aim of the Study
2.2. Design
2.3. Patient Selection and Classification
- Clopidogrel (for patients with ischemic stroke, myocardial infarction, or acute coronary syndrome).
- Fluoropyrimidines, tamoxifen, and irinotecan (for oncology patients starting chemotherapy).
- Azathioprine (for patients with autoimmune diseases requiring immunosuppressive therapy).
- Siponimod (for patients with secondary progressive multiple sclerosis—SPMS).
- Hospitalized patients required admission due to their underlying condition and the need for continuous monitoring, high-risk drug administration, or the management of severe immunosuppression or complications.
- Outpatients were clinically stable individuals who did not require hospitalization but needed PGx testing before treatment initiation. These included, for example, patients with stable cardiovascular conditions, oncology patients initiating chemotherapy as outpatients, or individuals with autoimmune diseases or neurological disorders who could be managed without inpatient care.
2.4. Organization and Planning
Structure and Functioning of the PGx Unit
- A nurse, responsible for collecting saliva samples from patients. Samples are collected at a PGx consultation within the Pharmacy Department for outpatients and at the hospital bedside for hospitalized patients. The nurse also ensures the proper identification and traceability of samples.
- A transport service, responsible for transferring samples from the hospital to an external laboratory.
- A laboratory team, which processes samples and generates genotyping results within 24 h, sending them to the hospital pharmacists.
- One pharmacist from the Pharmacy Department, responsible for interpreting genotypes, translating them into phenotypes, and providing personalized therapeutic recommendations to the medical departments requesting genetic testing. These recommendations are based on clinical practice guidelines, and, when necessary, discrepancies between different guidelines are harmonized using the available scientific evidence.
2.5. Sample and Data Collection
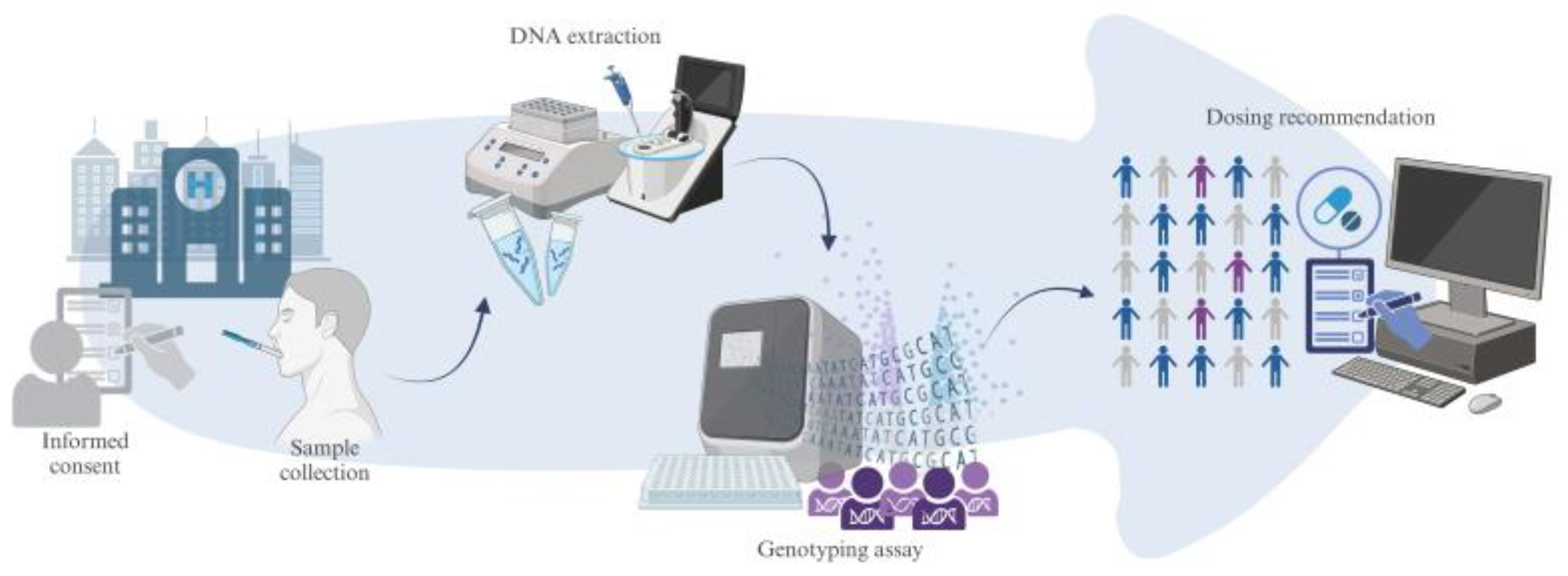
2.6. Genotyping and Results Reporting
2.7. Clinical Decision Support System
2.8. Data Management and Statistical Analysis
3. Results
3.1. Allelic and Genotypic Frequencies
3.2. Linkage Disequilibrium
3.3. Analysis of Haplotype Frequencies
4. Discussion
4.1. Relevance of Implemented Drug–Gene Interactions
4.2. Limitations
4.3. Clinical Implementation of Pharmacogenetics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-Fluorouracil |
| ADE | Adverse drug event |
| CPIC | Clinical Pharmacogenetics Implementation Consortium |
| CYP2C19 | Cytochrome P-450 family 2 subfamily C member 19 |
| CYP2D6 | Cytochrome P450 family 2 subfamily D member 6 |
| DNA | Deoxyribonucleic acid |
| DPYD | Dihydropyrimidine dehydrogenase |
| DPWG | Dutch Pharmacogenetics Working Group |
| EMA | European Medicines Agency |
| EUR | European |
| FDA | Federal Drug Administration |
| GOF | Gain of function |
| HET | Heterozygous genotype |
| HOM | Recessive homozygous genotype |
| H-W | Hardy–Weinberg |
| IBS | Iberian peninsula |
| ID | Identification number |
| IM | Intermediate metabolizer |
| LD | Linkage disequilibrium |
| LOF | Loss of function |
| MAF | Minor allele frequency |
| NM | Normal metabolizer |
| PGx | Pharmacogenetics |
| PM | Poor metabolizer |
| SNP | Single-nucleotide polymorphism |
| TPMT | Thiopurine methyltransferase |
| UGT1A1 | Uridine diphosphate glucuronosyltransferase 1 family, polypeptide A1 |
| UM | Ultrarapid metabolizer |
| WT | Wildtype |
Appendix A

{kind=link}
| CYP2D6 n = 23 | |||||||
| CYP2D6*3 | CYP2D6*4 | CYP2D6*5 | CYP2D6*6 | CYP2D6*9 | CYP2D6*10 | CYP2D6*41 | |
| CYP2D6*XN | 0.9286 −0.0295 0.8414 | 0.046 0.0175 0.9056 | 0.9286 −0.0295 0.8414 | 0.9286 −0.0295 0.8414 | 0.9774 −0.0444 0.7632 | 0.143 −0.0181 0.9023 | 0.9849 −0.0555 0.7068 |
| CYP2D6*3 | 0.987 −0.0825 0.5759 | 0.8571 −0.019 0.8972 | 0.8571 −0.019 0.8972 | 0.9286 −0.0295 0.8414 | 0.9881 −0.0875 0.5528 | 0.9524 −0.0375 0.7992 | |
| CYP2D6*4 | 0.987 −0.0825 0.5759 | 0.9959 0.2648 0.0725 | 0.9987 0.3798 0.01 | 0.9996 0.9433 0 | 0.9971 0.1477 0.3166 | ||
| CYP2D6*5 | 0.8571 −0.019 0.8972 | 0.9286 −0.0295 0.8414 | 0.9881 −0.0875 0.5528 | 0.9524 −0.0375 0.7992 | |||
| CYP2D6*6 | 0.9286 −0.0295 0.8414 | 0.9958 0.2499 0.0901 | 0.9524 −0.0375 0.7992 | ||||
| CYP2D6*9 | 0.9987 0.3584 0.0151 | 0.9849 −0.0555 0.7068 | |||||
| CYP2D6*10 | D´ r p-value | 0.9965 −0.1564 0.2889 | |||||
| DPYD n = 1502 | |||||||
| 1679T > G | 2846A > T | 1236G > A | |||||
| 1905+1G > A | 0.1447 0.0835 1 × 10−4 | 0.0517 0.0298 0.1718 | 0.0388 0.011 0.615 | ||||
| 1679T > G | 0.1422 0.0473 0.03 | 0.1306 0.0213 0.3288 | |||||
| 2846A > T | 0.3307 −0.0029 0.8942 | ||||||
| CYP2C19 n = 126 | |||||||
| CYP2C19*4 | CYP2C19*17 | ||||||
| CYP2C19*2 | 0.0351 0.0126 0.8412 | 0.9978 0.1689 0.0073 | |||||
| CYP2C19*4 | 0.9731 −0.0592 0.3476 | ||||||
| TPMT/NUDT15 n = 150 | |||||||
| TPMT*3B | TPMT*3C | NUDT15 | |||||
| TPMT*2 | 0.4195 −0.0047 0.9347 | 0.4678 −0.0055 0.9238 | 0.0147 0.0104 0.8572 | ||||
| TPMT*3B | 0.998 0.9539 0 | 0.8077 0.0129 0.8231 | |||||
| TPMT*3C | 0.8238 0.0138 0.8114 | ||||||
| CYP2C9 n = 32 | |||||||
| CYP2C9*3 | |||||||
| CYP2C9*2 | 0.421 0.269 0.0314 | ||||||
Appendix B
| CYP2D6 (n = 23) *star allele and Major allele > minor allele | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| *XN | *3 A > del | *4 G > A | *5 (del) | *6 T > del | *9 AAG > del | *10 C > T | *41 G > A | Total | Cumulative Frequency | |
| 1 | G | A | G | A | T | AAG | C | G | 0.6087 | 0.6087 |
| 2 | G | A | A | A | T | AAG | T | G | 0.1522 | 0.7609 |
| 3 | G | A | G | A | T | AAG | C | A | 0.058 | 0.8188 |
| 4 | G | A | A | A | T | del | T | G | 0.0435 | 0.8623 |
| 5 | xN | A | G | A | T | AAG | C | G | 0.029 | 0.8913 |
| 6 | G | A | A | A | del | AAG | T | G | 0.0217 | 0.913 |
| 7 | G | A | G | A | T | AAG | T | G | 0.0217 | 0.9348 |
| 8 | G | A | G | del | T | AAG | C | G | 0.0217 | 0.9565 |
| 9 | G | del | G | A | T | AAG | C | G | 0.0217 | 0.9783 |
| 10 | xN | A | A | A | T | AAG | T | G | 0.0145 | 0.9927 |
| 11 | G | A | A | A | T | AAG | T | A | 0.0073 | 1 |
| DPYD (n = 1502) Major nucleotide variation | ||||||||||
| 1905 + 1G > A | 1679 T > G | 2846 A > T | 1236 G > A | Total | Cumulative frequency | |||||
| 1 | C | A | T | C | 0.9762 | 0.9762 | ||||
| 2 | C | A | T | T | 0.0176 | 0.9938 | ||||
| 3 | C | A | A | C | 0.0043 | 0.9981 | ||||
| 4 | T | A | T | C | 0.0014 | 0.9995 | ||||
| 5 | C | C | T | C | 5 × 10−4 | 1 | ||||
| TPMT/NUDT15 (n = 150) *star allele and Major allele > minor allele | ||||||||||
| TPMT*2 G > C | TPMT*3B G > A | TPMT*3C A > G | NUDT15 C > T | Total | Cumulative frequency | |||||
| 1 | G | G | A | C | 0.95 | 0.95 | ||||
| 2 | G | A | G | C | 0.0367 | 0.9867 | ||||
| 3 | G | G | G | T | 0.0067 | 0.9933 | ||||
| 4 | C | G | A | C | 0.0033 | 0.9967 | ||||
| 5 | G | G | G | C | 0.0033 | 1 | ||||
| CYP2C9 (n = 32) *star allele and Major allele > minor allele | ||||||||||
| *2 C > T | *3 A > C | Total | Cumulative frequency | |||||||
| 1 | C | A | 0.7907 | 0.7907 | ||||||
| 2 | T | A | 0.1312 | 0.9219 | ||||||
| 3 | T | C | 0.0407 | 0.9625 | ||||||
| 4 | C | C | 0.0375 | 1 | ||||||
| CYP2C19 (n = 126) *star allele and Major allele > minor allele | ||||||||||
| *2 G > A | *4 A > G | *17 C > T | Total | Cumulative frequency | ||||||
| 1 | G | A | C | 0.6882 | 0.6882 | |||||
| 2 | G | A | T | 0.1865 | 0.8747 | |||||
| 3 | A | A | C | 0.1094 | 0.9841 | |||||
| 4 | G | G | C | 0.0142 | 0.9983 | |||||
References
- Kabbani, D.; Akika, R.; Wahid, A.; Daly, A.K.; Cascorbi, I.; Zgheib, N.K. Pharmacogenomics in practice: A review and implementation guide. Front. Pharmacol. 2023, 14, 1189976. [Google Scholar] [CrossRef] [PubMed]
- Shekhani, R.; Steinacher, L.; Swen, J.J.; Ingelman-Sundberg, M. Evaluation of Current Regulation and Guidelines of Pharmacogenomic Drug Labels: Opportunities for Improvements. Clin. Pharmacol. Ther. 2020, 107, 1240–1255. [Google Scholar] [CrossRef] [PubMed]
- FDA. Table of Pharmacogenetic Associations. 2020. Available online: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations (accessed on 22 January 2025).
- Relling, M.V.; Klein, T.E.; Gammal, R.S.; Whirl-Carrillo, M.; Hoffman, J.M.; Caudle, K.E. The Clinical Pharmacogenetics Implementation Consortium: 10 Years Later. Clin. Pharmacol. Ther. 2020, 107, 171–175. [Google Scholar] [CrossRef]
- Swen, J.; Wilting, I.; de Goede, A.; Grandia, L.; Mulder, H.; Touw, D.; de Boer, A.; Conemans, J.; Egberts, T.; Klungel, O.; et al. Pharmacogenetics: From bench to byte. Clin. Pharmacol. Ther. 2008, 84, 175. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Fragoulakis, V.; Bartsakoulia, M.; Díaz-Villamarín, X.; Chalikiopoulou, K.; Kehagia, K.; Ramos, J.G.S.; Martínez-González, L.J.; Gkotsi, M.; Katrali, E.; Skoufas, E.; et al. Cost-effectiveness analysis of pharmacogenomics-guided clopidogrel treatment in Spanish patients undergoing percutaneous coronary intervention. Pharmacogenomics J. 2019, 19, 438–445. [Google Scholar] [CrossRef]
- Turongkaravee, S.; Jittikoon, J.; Rochanathimoke, O.; Boyd, K.; Wu, O.; Chaikledkaew, U. Pharmacogenetic testing for adverse drug reaction prevention: Systematic review of economic evaluations and the appraisal of quality matters for clinical practice and implementation. BMC Health Serv. Res. 2021, 21, 1042. [Google Scholar] [CrossRef]
- Morris, S.A.; Alsaidi, A.T.; Verbyla, A.; Cruz, A.; Macfarlane, C.; Bauer, J.; Patel, J.N. Cost-effectiveness of pharmacogenetic testing for drugs with Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines: A systematic review. Clin. Pharmacol. Ther. 2022, 112, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Swanson, K.M.; Rojas, R.L.; Wang, Z.; Sauver, J.L.S.; Visscher, S.L.; Prokop, L.J.; Bielinski, S.J.; Wang, L.; Weinshilboum, R.; et al. Systematic review of the evidence on the cost-effectiveness of pharmacogenomics-guided treatment for cardiovascular diseases. Genet Med. 2020, 22, 475–486. [Google Scholar] [CrossRef]
- Hinderer, M.; Boeker, M.; Wagner, S.A.; Lablans, M.; Newe, S.; Hülsemann, J.L.; Neumaier, M.; Binder, H.; Renz, H.; Acker, T.; et al. Integrating clinical decision support systems for pharmacogenomic testing into clinical routine—A scoping review of designs of user-system interactions in recent system development. BMC Med. Inform. Decis. Mak. 2017, 17, 81. [Google Scholar] [CrossRef]
- Roncato, R.; Cin, L.D.; Mezzalira, S.; Comello, F.; De Mattia, E.; Bignucolo, A.; Giollo, L.; D’errico, S.; Gulotta, A.; Emili, L.; et al. FARMAPRICE: A Pharmacogenetic Clinical Decision Support System for Precise and Cost-Effective Therapy. Genes 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.L.; Rihal, C.S.; So, D.Y.; Rosenberg, Y.; Lennon, R.J.; Mathew, V.; Goodman, S.G.; Weinshilboum, R.M.; Wang, L.; Baudhuin, L.M.; et al. Clopidogrel Pharmacogenetics. Circ. Cardiovasc. Interv. 2019, 12, e007811. [Google Scholar] [CrossRef] [PubMed]
- Alkattan, A.; Alsalameen, E. Polymorphisms of genes related to phase-I metabolic enzymes affecting the clinical efficacy and safety of clopidogrel treatment. Expert Opin. Drug Metab. Toxicol. 2021, 17, 685–695. [Google Scholar] [CrossRef]
- CPIC. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Clopidogrel and CYP2C19. Available online: https://cpicpgx.org/guidelines/guideline-for-clopidogrel-and-cyp2c19/ (accessed on 8 March 2025).
- Tiroch, K.A.; Sibbing, D.; Koch, W.; Roosen-Runge, T.; Mehilli, J.; Schömig, A.; Kastrati, A. Protective effect of the CYP2C19 *17 polymorphism with increased activation of clopidogrel on cardiovascular events. Am. Heart J. 2010, 160, 506–512. [Google Scholar] [CrossRef]
- Sibbing, D.; Koch, W.; Gebhard, D.; Schuster, T.; Braun, S.; Stegherr, J.; Morath, T.; SchömIg, A.; von Beckerath, N.; Kastrati, A. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 2010, 121, 512–518. [Google Scholar] [CrossRef]
- Abdullah-Koolmees, H.; van Keulen, A.M.; Nijenhuis, M.; Deneer, V.H.M. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx Guidelines. Front. Pharmacol. 2020, 11, 595219. [Google Scholar] [CrossRef] [PubMed]
- Clinical Pharmacogenetics Implementation Consortium (CPIC). Guidelines for CYP2C19 and Voriconazole Therapy. Clin. Pharmacol. Ther. 2018, 103, 349. [Google Scholar] [CrossRef]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.P.J.M.; Van Schaik, R.H.N.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte–an update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef]
- Relling, M.V.; Schwab, M.; Whirl--Carrillo, M.; Suarez--Kurtz, G.; Pui, C.-H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; van der Wouden, C.H.; Nijenhuis, M.; Rhenen, M.H.C.-V.; de Boer-Veger, N.J.; Buunk, A.M.; Houwink, E.J.F.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. 2020, 28, 508–517. [Google Scholar] [CrossRef]
- Cecchin, E.; De Mattia, E.; Ecca, F.; Toffoli, G. Host genetic profiling to increase drug safety in colorectal cancer from discovery to implementation. Drug resistance updates: Reviews and commentaries in antimicrobial and anticancer chemotherapy. Drug Resist. Updates 2018, 39, 18–40. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Sangkuhl, K.; Guchelaar, H.; Schwab, M.; Province, M.; Whirl-Carrillo, M.; Symmans, W.F.; McLeod, H.L.; Ratain, M.J.; Zembutsu, H.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and Tamoxifen Therapy. Clin. Pharmacol. Ther. 2018, 103, 770–777. [Google Scholar] [CrossRef] [PubMed]
- (EMA) EMA. EMA Recommendations on DPD Testing Prior to Treatment with Fluorouracil, Capecitabine, Tegafur and Flucytosine. Available online: https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-and-flucytosine (accessed on 15 January 2025).
- Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Genome Aggregation Database Consortium; Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; et al. Author Correction: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2021, 590, E53. [Google Scholar] [CrossRef]
- van der Wouden, C.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.; Dávila-Fajardo, C.; Deneer, V.; Dolžan, V.; Ingelman-Sundberg, M.; Jönsson, S.; Karlsson, M.; et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 2017, 102, 152. [Google Scholar] [CrossRef]
- Freeman, B.; Smith, N.; Curtis, C.; Huckett, L.; Mill, J.; Craig, I. DNA from buccal swabs recruited by mail: Evaluation of storage effects on long-term stability and suitability for multiplex polymerase chain reaction genotyping. Behav. Genet. 2003, 33, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Martín, A.; Hernández, A.F.; Martínez-González, L.J.; González-Alzaga, B.; Rodríguez-Barranco, M.; López-Flores, I.; Aguilar-Garduno, C.; Lacasana, M. Polymorphisms of pesticide-metabolizing genes in children living in intensive farming communities. Chemosphere 2015, 139, 534–540. [Google Scholar] [CrossRef]
- Solé, X.; Guinó, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics. 2006, 22, 1928–1929. [Google Scholar] [CrossRef]
- Stewart, S.; Dodero-Anillo, J.M.; Guijarro-Eguinoa, J.; Arias, P.; Huertas, A.G.L.D.L.; Seco-Meseguer, E.; García-García, I.; García, E.R.; Rodríguez-Antolín, C.; Carcas, A.J.; et al. Advancing pharmacogenetic testing in a tertiary hospital: A retrospective analysis after 10 years of activity. Front. Pharmacol. 2023, 14, 1292416. [Google Scholar] [CrossRef]
- Pratt, V.M.; Cavallari, L.H.; Del Tredici, A.L.; Gaedigk, A.; Hachad, H.; Ji, Y.; Kalman, L.V.; Ly, R.C.; Moyer, A.M.; Scott, S.A.; et al. Recommendations for Clinical CYP2D6 Genotyping Allele Selection: A Joint Consensus Recommendation of the Association for Molecular Pathology, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, and the European Society for Pharmacogenomics and Personalized Therapy. J. Mol. Diagn. 2021, 23, 1047–1064. [Google Scholar] [CrossRef]
- Pratt, V.M.; Cavallari, L.H.; Fulmer, M.L.; Gaedigk, A.; Hachad, H.; Ji, Y.; Kalman, L.V.; Ly, R.C.; Moyer, A.M.; Scott, S.A.; et al. DPYD Genotyping Recommendations: A Joint Consensus Recommendation of the Association for Molecular Pathology, American College of Medical Genetics and Genomics, Clinical Pharmacogenetics Implementation Consortium, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, European Society for Pharmacogenomics and Personalized Therapy, Pharmacogenomics Knowledgebase, and Pharmacogene Variation Consortium. J. Mol. Diagn. 2024, 26, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- PharmGKB. Clinical Guideline Annotations for Azathioprine and TPMT. Available online: https://www.pharmgkb.org/chemical/PA10233/guidelineAnnotation/PA166104990 (accessed on 12 March 2025).
- Díaz-Villamarín, X.; Fernández-Varón, E.; Romero, M.C.R.; Callejas-Rubio, J.L.; Cabeza-Barrera, J.; Rodríguez-Nogales, A.; Gálvez, J.; Morón, R. Azathioprine dose tailoring based on pharmacogenetic information: Insights of clinical implementation. Biomed Pharmacother. 2023, 168, 115706. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Sangkuhl, K.; Guchelaar, H.; Schwab, M.; Province, M.; Whirl-Carrillo, M.; Symmans, W.F.; McLeod, H.L.; Ratain, M.J.; Zembutsu, H.; et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 2021, 32, 1475–1495. [Google Scholar] [CrossRef]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.P.J.M.; Van Schaik, R.H.N.; Schalekamp, T.; Touw, D.J.; et al. Systemic Treatment of Patients With Metastatic Breast Cancer: ASCO Resource-Stratified Guideline. JCO Glob. Oncol. 2024, 10, e2300285. [Google Scholar] [CrossRef]
- Díaz-Villamarín, X.; Piñar-Morales, R.; Barrero-Hernández, F.J.; Antúnez-Rodríguez, A.; Cabeza-Barrera, J.; Morón-Romero, R. Pharmacogenetics of siponimod: A systematic review. Biomed. Pharmacother. 2022, 153, 113536. [Google Scholar] [CrossRef]
- Del Tredici, A.L.; Malhotra, A.; Dedek, M.; Espin, F.; Roach, D.; Zhu, G.-D.; Voland, J.; Moreno, T.A. Frequency of CYP2D6 Alleles Including Structural Variants in the United States. Front Pharmacol. 2018, 9, 305. [Google Scholar] [CrossRef]
- Nunez-Torres, R.; Pita, G.; Peña-Chilet, M.; López-López, D.; Zamora, J.; Roldán, G.; Herráez, B.; Álvarez, N.; Alonso, M.R.; Dopazo, J.; et al. A Comprehensive Analysis of 21 Actionable Pharmacogenes in the Spanish Population: From Genetic Characterisation to Clinical Impact. Pharmaceutics 2023, 15, 1286. [Google Scholar] [CrossRef]
- Cecchin, E.; Roncato, R.; Guchelaar, H.J.; Toffoli, G. Ubiquitous Pharmacogenomics (U-PGx): The Time for Implementation is Now. An Horizon2020 Program to Drive Pharmacogenomics into Clinical Practice. Curr. Pharm. Biotechnol. 2017, 18, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.-C.; et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: An open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 2023, 401, 347–356. [Google Scholar] [CrossRef] [PubMed]
- de Sanidad, M. Cartera de Servicios Comunes del Sistema Nacional de Salud: Actualización. 2022. Available online: https://www.sanidad.gob.es/organizacion/consejoInterterri/docs/1553.pdf (accessed on 25 January 2025).
- Díaz-Villamarín, X.; Martínez-Pérez, M.; Nieto-Sánchez, M.T.; Ruiz-Tueros, G.; Fernández-Varón, E.; Torres-García, A.; González Astorga, B.; Blancas, I.; Iáñez, A.J.; Cabeza-Barrera, J.; et al. Novel Genetic Variants Explaining Severe Adverse Drug Events after Clinical Implementation of DPYD Genotype-Guided Therapy with Fluoropyrimidines: An Observational Study. Pharmaceutics 2024, 16, 956. [Google Scholar] [CrossRef] [PubMed]
- Llerena, A.; Peñas-Lledó, E.; de Andrés, F.; Mata-Martín, C.; Sánchez, C.L.; Pijierro, A.; Cobaleda, J. Clinical implementation of pharmacogenetics and personalized drug prescription based on e-health: The MedeA initiative. Drug Metab. Pers. Ther. 2020, 35, 20200143. [Google Scholar] [CrossRef]
| Drug | Gene | Star Allele | Major Nucleotide Variation | dbSNP RS ID | Phenotypes |
|---|---|---|---|---|---|
| Clopidogrel | CYP2C19 | *2 | 681G > A | rs4244285 | PM/IM/NM/RM/UM |
| *3 | 636G > A | rs4986893 | |||
| *4A/B | 1A > G | rs28399504 | |||
| *17 | −806C > T3 | rs12248560 | |||
| Azathioprine | TPMT | *2 | 238G > C | rs1800462 | PM/IM/NM |
| *3B | 460G > A | rs1800460 | |||
| *3C | 719A > G | rs1142345 | |||
| NUDT15 | *3 | 7973C > T | rs116855232 | ||
| Capecitabine/5-FU | DPYD | *2A | IVS14 + 1G > A (1905 + 1G > A) | rs3918290 | GAS: 0;0.5;1;1.5;2 |
| *13 | 1679T > G | rs55886062 | |||
| - | 2846A > T | rs67376798 | |||
| - | 1236G > A | rs56038477 | |||
| Irinotecan | UGT1A1 | *6 | 211G > A | rs4148323 | PM/NM |
| *27 | 686C > A | rs35350960 | |||
| *28/*37 | TA6 > TA7 or TA8 | rs3064744 | |||
| Tamoxifen | CYP2D6 | *xN | Gene multiplication | - | PM/IM/NM/UM |
| *3 | 2549delA | rs35742686 | |||
| *4 | 1846G > A | rs3892097 | |||
| *5 | Gene deletion | - | |||
| *6 | 1707delT | rs5030655 | |||
| *8 | 1758G > T | rs5030865 | |||
| *9 | 2615delAAG | rs5030656 | |||
| *10 | 100C > T | rs1065852 | |||
| *14A/B | 1758G > A | rs5030865 | |||
| *17 | 1023C > T | rs28371706 | |||
| *41 | 2988G > A | rs28371725 | |||
| Siponimod | CYP2C9 | *2 | 3608C > T | rs1799853 | PM/IM/NM |
| *3 | 42614A > C | rs1057910 |
| Phenotypes n (%) | Genotypes GENE* Carried Star Allele = Number of Patients | Therapeutic Recommendation |
|---|---|---|
| Clopidogrel: CYP2C19 (n = 126) | ||
| UM: 8 (6.35) | CYP2C19*17/*17 = 8 | NO 76.19% |
| NM: 64 (50.79) | CYP2C19*1/*1 = 64 | |
| RM: 24 (19.05) | CYP2C19*1/*17 = 24 | |
| IM: 28 (22.22) | CYP2C19*1/*2 = 18; *1/*4 = 3; *2/*17 = 7 | YES 23.81% |
| PM: 2 (1.59) | CYP2C19*2/*2 = 1; *2/*4 = 1 | |
| Azathioprine: TPMT and NUDT15 (n = 150) | ||
| NM: 135 (90.00) | TPMT*1/*1 = 135; NUDT15*1/*1 = 135 | NO 90.00% |
| IM: 15 (10.00) | TPMT*3B/*3C (TPMT*1/*3A) =11; *1/*2 = 1; *1/*3C = 1. NUDT15*1/*3 = 2 | YES 10.00% |
| PM: 0 (0) | - | |
| Capecitabine/5-FU:DPYD (n = 1052) | ||
| GAS 2: 1005 (95.53) | DPYD*1/*1 = 1005 | NO 95.53% |
| GAS 1.5: 46 (4.37) | DPYD*1/rs56038477 = 37; *1/rs67376798 = 9 | YES 4.47% |
| GAS 1: 1 (0.01) | DPYD*1/*13 = 1 | |
| Irinotecan:UGT1A1 (n = 150) | ||
| NM: 65 (43.33) | UGT1A1*1/*1 = 65 | NO 84.67% |
| IM: 62 (41.33) | UGT1A1*1/*28 = 62 | |
| PM: 23 (15.33) | UGT1A1*28/*28 = 23 | YES 15.33% |
| Tamoxifen:CYP2D6 (n = 23) | ||
| NM: 9 (39.13) | CYP2D6*1/*1= 6; *1/*41 = 2; *1/*10 = 1; | NO 43.48% |
| UM: 1 (4.35) | CYP2D6*1/*xN = 1 | |
| IM: 13 (56.52) | CYP2D6*1/*5 = 1; *1/*3 = 1; *4/*10 = 6; *4/*xN/*10 = 1; *4/*10/*6 = 1; *4/*10/*9 = 2; *4/*10/*41 = 1 | YES 56.52% |
| PM: 0 (0) | - | |
| Siponimod:CYP2C9 (n = 32) | ||
| NM: 20 (62.5) | CYP2C9*1/*1 = 20 | NO 87.50% |
| IM: 8 (25.0) | CYP2C9*1/*2 = 8 | |
| PM: 4 (12.5) | CYP2C9*2/*3 = 4 | YES 12.50% |
| TOTAL: n = 1533 | ||
| n = 1401 (91.39) | - | NO 91.39% |
| n = 132 (8.61) | - | YES 8.61% |
| Requested PGx Test n = 1533 | SNP (star allele) | Major Nucleotide Variation | dbSNP RS ID | Our Population n = 1567 ⴕ | IBS n = 107 | EUR n = 503 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wt n (%) | Het n (%) | Hom n (%) | MAF | p-Value (H-W) | p-Value | p-Value | ||||
| Clopidogrel n = 126 | CYP2C19*2 | 681G > A | rs4244285 | 99 (78.57) | 26 (20.63) | 1 (0.80) | 0.111 | 1 | 0.275 | 0.185 |
| CYP2C19*3 | 636G > A | rs4986893 | 126 (100) | 0 (0) | 0 (0) | 0 | - | 1 * | 1 * | |
| CYP2C19*4A/B | 1A > G | rs28399504 | 122 (96.82) | 4 (3.18) | 0 (0) | 0.016 | 1 | 0.381 | 0.007 | |
| CYP2C19*17 | −806C > T3 | rs12248560 | 87 (69.05) | 31 (24.60) | 8 (6.35) | 0.187 | 0.039 | 0.444 | 0.200 | |
| Azathioprine n = 150 | TPMT*2 | 238G > C | rs1800462 | 149 (99.33) | 1 (0.67) | 0 (0) | 0.003 | 1 | 0.573 * | 1 * |
| TPMT*3B | 460G > A | rs1800460 | 139 (92.67) | 11 (7.33) | 0 (0) | 0.037 | 1 | 0.755 | 0.430 | |
| TPMT*3C | 719A > G | rs1142345 | 138 (92.00) | 12 (8.00) | 0 (0) | 0.04 | 1 | 0.908 | 0.330 | |
| NUDT15*3 | 7973C > T | rs116855232 | 148 (98.67) | 2 (1.33) | 0 (0) | 0.007 | 1 | 0.513 * | 0.228 * | |
| Capecitabine/5-FU n = 1052 | DPYD*2A | IVS14 + 1G > A | rs3918290 | 1049 (99.71) | 3 (0.29) | 0 (0) | 0.001 | 1 | 1 * | 0.122 * |
| DPYD*13 | 1679T > G | rs55886062 | 1051 (99.90) | 1 (0.10) | 0 (0) | 0.001 | 1 | 1 * | 0.542 * | |
| - | 2846A > T | rs67376798 | 1043 (99.14) | 9 (0.86) | 0 (0) | 0.004 | 1 | 1 * | 0.328 | |
| - | 1236G > A | rs56038477 | 1015 (96.48) | 37 (3.52) | 0 (0) | 0.018 | 1 | 0.907 | 0.721 | |
| Irinotecan n = 150 | UGT1A1*6 | 211G > A | rs4148323 | 150 (100) | 0 (0) | 0 (0) | 0 | - | 1 * | 0.604 * |
| UGT1A1*27 | 686C > A | rs35350960 | 150 (100) | 0 (0) | 0 (0) | 0 | - | 1 * | 1 * | |
| UGT1A1*28/*37 | TA6 > TA7 or TA8 | rs3064744 | 65 (43.34) | 62 (41.33) | 23 (15.33) | 0.36 | 0.22 | NA | 0.147 | |
| Tamoxifen n = 23 | CYP2D6*xN | Multiplication | - | 21 (87.5) | 2 (12.5) | 0 (0) | 0.043 | 1 | NA | NA |
| CYP2D6*3 | 2549delA | rs35742686 | 22 (91.66) | 1 (8.33) | 0 (0) | 0.022 | 1 | 0.449 * | 0.594 * | |
| CYP2D6*4 | 1846G > A | rs3892097 | 12 (52.17) | 11 (47.83) | 0 (0) | 0.239 | 0.28 | 0.115 | 0.366 | |
| CYP2D6*5 | Deletion | - | 22 (91.66) | 1 (8.33) | 0 (0) | 0.022 | 1 | NA | NA | |
| CYP2D6*6 | 1707delT | rs5030655 | 22 (91.66) | 1 (8.33) | 0 (0) | 0.022 | 1 | 0.323 * | 0.613 * | |
| CYP2D6*8 | 1758G > T | rs5030865 | 23 (100) | 0 (0) | 0 (0) | 0 | - | 1 * | 1 * | |
| CYP2D6*9 | 2615delAAG | rs5030656 | 21 (87.5) | 2 (12.5) | 0 (0) | 0.043 | 1 | 0.359 * | 0.349 * | |
| CYP2D6*10 | 100C > T | rs1065852 | 11 (47.83) | 12 (52.17) | 0 (0) | 0.239 | 0.27 | 0.293 | 0.538 | |
| CYP2D6*14A/B | 1758G > A | rs5030865 | 23 (100) | 0 (0) | 0 (0) | 0 | - | 1 * | 1 * | |
| CYP2D6*17 | 1023C > T | rs28371706 | 23 (100) | 0 (0) | 0 (0) | 0 | - | 1 * | 1 * | |
| CYP2D6*41 | 2988G > A | rs28371725 | 20 (86.95) | 3 (13.05) | 0 (0) | 0.065 | - | 0.775 * | 0.793 * | |
| Siponimod n = 32 | CYP2C9*2 | 3608C > T | rs1799853 | 21 (65.62) | 11 (34.38) | 0 (0) | 0.172 | 0.56 | 0.530 | 0.267 |
| CYP2C9*3 | 42614A > C | rs1057910 | 28 (87.5) | 3 (9.38) | 1 (3.12) | 0.078 | 0.15 | 0.879 | 0.868 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Villamarín, X.; Martínez-Pérez, M.; Nieto-Sánchez, M.T.; Fernández-Varón, E.; Torres-García, A.; Blancas, I.; Cabeza-Barrera, J.; Morón, R. Clinical Pharmacogenetics: Results After Implementation of Preemptive Tests in Daily Routine. J. Pers. Med. 2025, 15, 245. https://doi.org/10.3390/jpm15060245
Díaz-Villamarín X, Martínez-Pérez M, Nieto-Sánchez MT, Fernández-Varón E, Torres-García A, Blancas I, Cabeza-Barrera J, Morón R. Clinical Pharmacogenetics: Results After Implementation of Preemptive Tests in Daily Routine. Journal of Personalized Medicine. 2025; 15(6):245. https://doi.org/10.3390/jpm15060245
Chicago/Turabian StyleDíaz-Villamarín, Xando, María Martínez-Pérez, María Teresa Nieto-Sánchez, Emilio Fernández-Varón, Alicia Torres-García, Isabel Blancas, José Cabeza-Barrera, and Rocío Morón. 2025. "Clinical Pharmacogenetics: Results After Implementation of Preemptive Tests in Daily Routine" Journal of Personalized Medicine 15, no. 6: 245. https://doi.org/10.3390/jpm15060245
APA StyleDíaz-Villamarín, X., Martínez-Pérez, M., Nieto-Sánchez, M. T., Fernández-Varón, E., Torres-García, A., Blancas, I., Cabeza-Barrera, J., & Morón, R. (2025). Clinical Pharmacogenetics: Results After Implementation of Preemptive Tests in Daily Routine. Journal of Personalized Medicine, 15(6), 245. https://doi.org/10.3390/jpm15060245