
Personalizing DNA Cancer Vaccines

 , , ,
, , ,
Abstract
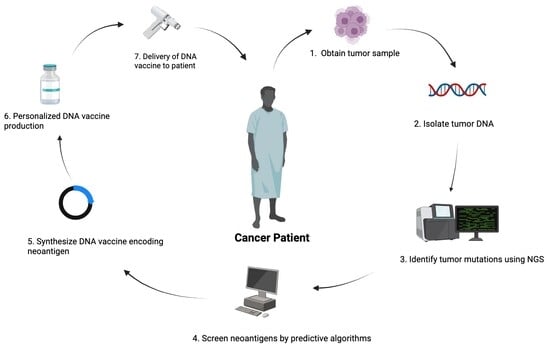
1. Introduction
2. Classes of Tumor Antigens
2.1. Shared Antigens
2.2. Personalized Neoantigens
3. Identification of Neoantigens
3.1. Neoantigen Identification and Sources
3.2. Antigen Processing, MHC Presentation, and Immunogenicity Prediction
3.3. Prioritization and Validation of Candidate Neoantigens
4. DNA-Based Vaccine Platforms
Personalized DNA Vaccine for Cancer Immunotherapy
5. Limitations in DNA Vaccine Effectiveness and Solutions
6. Considerations in Clinical Trials
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen-presenting cells |
| COVID-19 | Coronavirus disease-2019 |
| CPTAC | Clinical Proteomic Tumor Analysis Consortium |
| CTA | Cancer-testis antigen |
| CTL | Cytotoxic lymphocytes |
| DC | Dendritic cells |
| DNA | Deoxyribonucleic acid |
| EBV | Epstein–Barr virus |
| EGFR | Epidermal growth factor receptor |
| ELISpot | Enzyme-linked immunosorbent spot assay |
| FDA | Food and Drug Administration |
| FFPE | Formalin-fixed and paraffin-embedded |
| GM-CSF | Granulocyte macrophage-colony stimulating factor |
| GTEx | Genotype-Tissue Expression project |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HER | Human epidermal growth factor receptor |
| HLA | Human leukocyte antigen |
| HPV | Human papillomavirus |
| hTERT | Human telomerase reverse transcriptase |
| ICI | Immune checkpoint inhibitors |
| IDO | Indoleamine 2,3-dioxygenase |
| IFN-γ | Interferon gamma |
| IGFBP2 | Insulin-like growth factor binding protein 2 |
| IL | Interleukin |
| KRAS | Kirsten rat sarcoma virus |
| LNP | Lipid nanoparticles |
| MAGE | Melanoma antigen gene |
| MHC class I | Major histocompatibility complex class I |
| MHC class II | Major histocompatibility complex class II |
| mRNA | Messenger ribonucleic acid |
| NGS | Next-generation sequencing |
| NY-ESO-1 | New York esophageal squamous cell carcinoma 1 |
| PBMC | Peripheral blood mononuclear cells |
| PD-L1 | Programmed death-ligand 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| pMHC | peptide-MHC complex |
| PSA | Prostate-specific antigen |
| PSMA | Prostate-specific membrane antigen |
| RFS | Recurrence-free survival |
| RiboSeq | Ribosome profiling |
| RNA-seq | RNA sequencing |
| SNV | Single nucleotide variations |
| STING | Stimulator of interferon genes |
| TAA | Tumor-associated antigen |
| TCGA | The Cancer Genome Atlas |
| TCR | T cell receptor |
| TIL | Tumor infiltrating lymphocytes |
| TLR | Toll-like receptor |
| TMB | Tumor mutational burden |
| TNF | Tumor necrosis factor |
| TP53 | Tumor protein p53 |
| TSA | Tumor-specific antigen |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| WT1 | Wilms tumor 1 |
References
- Ottensmeier, C.H.H.; Delord, J.P.; Lalanne, A.; Lantz, O.; Jamet, C.; Tavernaro, A.; Brandely-Talbot, M.; Grellier, B.; Bastien, B.; Makhloufi, H.; et al. Safety and immunogenicity of TG4050: A personalized cancer vaccine in head and neck carcinoma. J. Clin. Oncol. 2023, 41, 6082. [Google Scholar] [CrossRef]
- Aggarwal, C.; Ben-Shachar, R.; Gao, Y.; Hyun, S.W.; Rivers, Z.; Epstein, C.; Kaneva, K.; Sangli, C.; Nimeiri, H.; Patel, J. Assessment of Tumor Mutational Burden and Outcomes in Patients with Diverse Advanced Cancers Treated with Immunotherapy. JAMA Netw. Open 2023, 6, e2311181. [Google Scholar] [CrossRef]
- Quintanilha, J.C.F.; Storandt, M.H.; Graf, R.P.; Li, G.; Keller, R.; Lin, D.I.; Ross, J.S.; Huang, R.S.P.; Schrock, A.B.; Oxnard, G.R.; et al. Tumor Mutational Burden in Real-World Patients with Pancreatic Cancer: Genomic Alterations and Predictive Value for Immune Checkpoint Inhibitor Effectiveness. JCO Precis Oncol. 2023, 7, e2300092. [Google Scholar] [CrossRef]
- Zhang, X.; Goedegebuure, S.P.; Chen, M.Y.; Mishra, R.; Zhang, F.; Yu, Y.Y.; Singhal, K.; Li, L.; Gao, F.; Myers, N.B.; et al. Neoantigen DNA vaccines are safe, feasible, and induce neoantigen-specific immune responses in triple-negative breast cancer patients. Genome Med. 2024, 16, 131. [Google Scholar] [CrossRef]
- Yarchoan, M.; Gane, E.J.; Marron, T.U.; Perales-Linares, R.; Yan, J.; Cooch, N.; Shu, D.H.; Fertig, E.J.; Kagohara, L.T.; Bartha, G.; et al. Personalized neoantigen vaccine and pembrolizumab in advanced hepatocellular carcinoma: A phase 1/2 trial. Nat. Med. 2024, 30, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Toyoshima, Y.; Nakamura, Y. Personalized immunotherapy in cancer precision medicine. Cancer Biol. Med. 2021, 18, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Harrington, K.J.; Zocca, M.B.; Ehrnrooth, E.; Cohen, E.E.W. The Changing Landscape of Therapeutic Cancer Vaccines-Novel Platforms and Neoantigen Identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges, and prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. npj Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Papachristofilou, A.; Hipp, M.M.; Klinkhardt, U.; Früh, M.; Sebastian, M.; Weiss, C.; Pless, M.; Cathomas, R.; Hilbe, W.; Pall, G.; et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 38. [Google Scholar] [CrossRef]
- Gnjatic, S.; Altorki, N.K.; Tang, D.N.; Tu, S.M.; Kundra, V.; Ritter, G.; Old, L.J.; Logothetis, C.J.; Sharma, P. NY-ESO-1 DNA vaccine induces T-cell responses that are suppressed by regulatory T cells. Clin. Cancer Res. 2009, 15, 2130–2139. [Google Scholar] [CrossRef]
- Xue, W.; Metheringham, R.L.; Brentville, V.A.; Gunn, B.; Symonds, P.; Yagita, H.; Ramage, J.M.; Durrant, L.G. SCIB2, an antibody DNA vaccine encoding NY-ESO-1 epitopes, induces potent antitumor immunity which is further enhanced by checkpoint blockade. Oncoimmunology 2016, 5, e1169353. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Hannen, R.; Bartsch, J.W. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018, 592, 2023–2031. [Google Scholar] [CrossRef]
- Teixeira, L.; Medioni, J.; Garibal, J.; Adotevi, O.; Doucet, L.; Durey, M.D.; Ghrieb, Z.; Kiladjian, J.J.; Brizard, M.; Laheurte, C.; et al. A First-in-Human Phase I Study of INVAC-1, an Optimized Human Telomerase DNA Vaccine in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 588–597. [Google Scholar] [CrossRef]
- Jo, J.H.; Kim, Y.T.; Choi, H.S.; Kim, H.G.; Lee, H.S.; Choi, Y.W.; Kim, D.U.; Lee, K.H.; Kim, E.J.; Han, J.H.; et al. Efficacy of GV1001 with gemcitabine/capecitabine in previously untreated patients with advanced pancreatic ductal adenocarcinoma having high serum eotaxin levels (KG4/2015): An open-label, randomised, Phase 3 trial. Br. J. Cancer 2024, 130, 43–52. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Kraynyak, K.A.; Shields, A.F.; McRee, A.J.; Johnson, J.M.; Sun, W.; Chintakuntlawar, A.V.; Pawlicki, J.; Sylvester, A.J.; McMullan, T.; et al. Phase 1 study of safety, tolerability and immunogenicity of the human telomerase (hTERT)-encoded DNA plasmids INO-1400 and INO-1401 with or without IL-12 DNA plasmid INO-9012 in adult patients with solid tumors. J. Immunother. Cancer 2021, 9, e003019. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brem, S.; Desai, A.S.; Bagley, S.J.; Kurz, S.C.; De La Fuente, M.I.; Nagpal, S.; Welch, M.R.; Hormigo, A.; Forsyth, P.A.J.; et al. Intramuscular (IM) INO-5401+INO-9012 with electroporation (EP) in combination with cemiplimab (REGN2810) in newly diagnosed glioblastoma. J. Clin. Oncol. 2022, 40, 2004. [Google Scholar] [CrossRef]
- Madan, R.A.; Arlen, P.M.; Mohebtash, M.; Hodge, J.W.; Gulley, J.L. Prostvac-VF: A vector-based vaccine targeting PSA in prostate cancer. Expert Opin. Investig. Drugs 2009, 18, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef]
- Linch, M.; Papai, Z.; Takacs, I.; Imedio, E.R.; Kühnle, M.-C.; Derhovanessian, E.; Vogler, I.; Renken, S.; Graham, P.; Sahin, U.; et al. 421 A first-in-human (FIH) phase I/IIa clinical trial assessing a ribonucleic acid lipoplex (RNA-LPX) encoding shared tumor antigens for immunotherapy of prostate cancer; preliminary analysis of PRO-MERIT. J. Immunother. Cancer 2021, 9, A451. [Google Scholar] [CrossRef]
- Patel, P.H.; Kockler, D.R. Sipuleucel-T: A vaccine for metastatic, asymptomatic, androgen-independent prostate cancer. Ann. Pharmacother. 2008, 42, 91–98. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Crosby, E.J.; Gwin, W.; Blackwell, K.; Marcom, P.K.; Chang, S.; Maecker, H.T.; Broadwater, G.; Hyslop, T.; Kim, S.; Rogatko, A.; et al. Vaccine-Induced Memory CD8+ T Cells Provide Clinical Benefit in HER2 Expressing Breast Cancer: A Mouse to Human Translational Study. Clin. Cancer Res. 2019, 25, 2725–2736. [Google Scholar] [CrossRef]
- Kang, J.; Park, H.-H.; Choi, J.H.; Lim, J.; Jang, S.-Y.; Kim, M.-A.; Park, M.-K.; Park, Y.-H.; Lim, S.; Chung, C.-W.; et al. 897 AST-301, a pDNA-based cancer vaccine encoding HER2-ICD, enhances anti-tumor effect of HER2-ADC in a HER2-expressed gastric cancer xenograft model. J. Immunother. Cancer 2023, 11, A999. [Google Scholar] [CrossRef]
- Lim, J.; Park, H.-H.; Choi, J.K.; Choi, J.H.; Kang, J.; Jang, S.-Y.; Kim, M.-A.; Park, M.-K.; Disis, M.L.; Joung, E.; et al. 817 AST-201 (pUMVC3-hIGFBP2 N-terminus) demonstrates anti-tumor effect in an ovarian cancer mouse model. J. Immunother. Cancer 2023, 11, A915. [Google Scholar] [CrossRef]
- Han, H.S.; Wesolowski, R.; Fisher, C.; Gandhi, S.; Gwin, W.R.; Kowzun, M.J.; Gogineni, K.; Liu, H.; Costa, R.L.; Guerrero, J.; et al. A multicenter phase II study of vaccines to prevent recurrence in patients with HER-2-positive breast cancer. J. Clin. Oncol. 2023, 41, 532. [Google Scholar] [CrossRef]
- Gwin, W.R.; Kuano, K.; Childs, J.; Symonds, L.K.; Coveler, A.L.; Liao, J.B.; Vinayak, S.; Disis, M.L. A phase II study of concurrent WOKVAC vaccination with neoadjuvant chemotherapy and HER2-targeted monoclonal antibody therapy. J. Clin. Oncol. 2023, 41, TPS636. [Google Scholar] [CrossRef]
- Pan, R.Y.; Chung, W.H.; Chu, M.T.; Chen, S.J.; Chen, H.C.; Zheng, L.; Hung, S.I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol. Res. 2018, 2018, 4325874. [Google Scholar] [CrossRef]
- Romero, P.; Banchereau, J.; Bhardwaj, N.; Cockett, M.; Disis, M.L.; Dranoff, G.; Gilboa, E.; Hammond, S.A.; Hershberg, R.; Korman, A.J.; et al. The Human Vaccines Project: A roadmap for cancer vaccine development. Sci. Transl. Med. 2016, 8, 334ps9. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, N.; Treat, B.; Duensing, S. The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol. Cancer 2011, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Albert, E.; Laimins, L. Regulation of the Human Papillomavirus Life Cycle by DNA Damage Repair Pathways and Epigenetic Factors. Viruses 2020, 12, 744. [Google Scholar] [CrossRef] [PubMed]
- Sofiani, V.H.; Veisi, P.; Rukerd, M.R.Z.; Ghazi, R.; Nakhaie, M. The complexity of human papilloma virus in cancers: A narrative review. Infect. Agent Cancer 2023, 18, 13. [Google Scholar] [CrossRef]
- Klinghammer, K.; Saba, N.F.; Castelluci, E.; Colevas, A.D.; Rutkowski, T.; Greil, R.; Thurner, D.; Müller-Richter, U.; Di Giacomo, A.M.; Grewal, J.; et al. 155P BNT113 + pembrolizumab as first-line treatment in patients with unresectable recurrent/metastatic HNSCC: Preliminary safety data from AHEAD-MERIT. Immuno. Oncol. Technol. 2022, 16, 100267. [Google Scholar] [CrossRef]
- Youn, J.W.; Hur, S.Y.; Woo, J.W.; Kim, Y.M.; Lim, M.C.; Park, S.Y.; Seo, S.S.; No, J.H.; Kim, B.G.; Lee, J.K.; et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: Interim results of a single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1653–1660. [Google Scholar] [CrossRef]
- Gibson, M.K.; Savvides, P.; Worden, F.; Heimann-Nichols, E.; Wu, T.; Roden, R.; Chang, Y.-N. 677 Phase II trial assessing safety, efficacy and immune correlates of heterologous prime-boost with pBI-11 (IM) and TA-HPV (IM) plus pembrolizumab for advanced, PD-L1 CPS≥1, hrHPV+ Oropharyngeal cancer. J. Immunother. Cancer 2023, 11, A768. [Google Scholar] [CrossRef]
- Peng, S.; Qiu, J.; Yang, A.; Yang, B.; Jeang, J.; Wang, J.W.; Chang, Y.N.; Brayton, C.; Roden, R.B.S.; Hung, C.F.; et al. Optimization of heterologous DNA-prime, protein boost regimens and site of vaccination to enhance therapeutic immunity against human papillomavirus-associated disease. Cell Biosci. 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Einstein, M.H.; Roden, R.B.S.; Ferrall, L.; Akin, M.; Blomer, A.; Wu, T.C.; Chang, Y.N. Safety Run-in of Intramuscular pNGVL4a-Sig/E7(detox)/HSP70 DNA and TA-CIN Protein Vaccination as Treatment for HPV16+ ASC-US, ASC-H, or LSIL/CIN1. Cancer Prev. Res. (Phila) 2023, 16, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hillemanns, P.; Denecke, A.; Woelber, L.; Böhmer, G.; Jentschke, M.; Schjetne, K.W.; Bruins Slot, K.M.H.; Fredriksen, A.B. A Therapeutic Antigen-Presenting Cell-Targeting DNA Vaccine VB10.16 in HPV16-Positive High-Grade Cervical Intraepithelial Neoplasia: Results from a Phase I/IIa Trial. Clin. Cancer Res. 2022, 28, 4885–4892. [Google Scholar] [CrossRef]
- Bhuyan, P.K.; Dallas, M.; Kraynyak, K.; Herring, T.; Morrow, M.; Boyer, J.; Duff, S.; Kim, J.; Weiner, D.B. Durability of response to VGX-3100 treatment of HPV16/18 positive cervical HSIL. Hum. Vaccin. Immunother. 2021, 17, 1288–1293. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Gupta, R.G.; Li, F.; Roszik, J.; Lizée, G. Exploiting Tumor Neoantigens to Target Cancer Evolution: Current Challenges and Promising Therapeutic Approaches. Cancer Discov. 2021, 11, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; McMichael, J.; Becker-Hapak, M.; Onyeador, O.C.; Buchli, R.; McClain, E.; Pence, P.; Supabphol, S.; Richters, M.M.; Basu, A.; et al. Computational prediction of MHC anchor locations guides neoantigen identification and prioritization. Sci. Immunol. 2023, 8, eabg2200. [Google Scholar] [CrossRef]
- Vilimas, T. Measuring Tumor Mutational Burden Using Whole-Exome Sequencing. Methods Mol. Biol. 2020, 2055, 63–91. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Vazquez, M.; Finotello, F.; Lepore, R.; Porta, E.; Hundal, J.; Amengual-Rigo, P.; Ng, C.K.Y.; Valencia, A.; Carrillo, J.; et al. Neoantigen prediction and computational perspectives towards clinical benefit: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 978–990. [Google Scholar] [CrossRef]
- Shi, Y.; Jing, B.; Xi, R. Comprehensive analysis of neoantigens derived from structural variation across whole genomes from 2528 tumors. Genome Biol. 2023, 24, 169. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Antón, L.C.; Bennink, J.R. Defective ribosomal products (DRiPs): A major source of antigenic peptides for MHC class I molecules? J. Immunol. 1996, 157, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Hansen, U.K.; Ramskov, S.; Bjerregaard, A.M.; Borch, A.; Andersen, R.; Draghi, A.; Donia, M.; Bentzen, A.K.; Marquard, A.M.; Szallasi, Z.; et al. Tumor-Infiltrating T Cells From Clear Cell Renal Cell Carcinoma Patients Recognize Neoepitopes Derived From Point and Frameshift Mutations. Front. Immunol. 2020, 11, 373. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Wei, Z.; Zhou, C.; Zhang, Z.; Guan, M.; Zhang, C.; Liu, Z.; Liu, Q. The Landscape of Tumor Fusion Neoantigens: A Pan-Cancer Analysis. iScience 2019, 21, 249–260. [Google Scholar] [CrossRef]
- Romanish, M.T.; Cohen, C.J.; Mager, D.L. Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin. Cancer Biol. 2010, 20, 246–253. [Google Scholar] [CrossRef]
- Schliehe, C.; Bitzer, A.; van den Broek, M.; Groettrup, M. Stable antigen is most effective for eliciting CD8+ T-cell responses after DNA vaccination and infection with recombinant vaccinia virus in vivo. J. Virol. 2012, 86, 9782–9793. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Sacks, N.J.; Schenkel, J.M.; Ely, Z.A.; Smith, O.; Hauck, H.; Jaeger, A.M.; Zhang, D.; Backlund, C.M.; Beytagh, M.C.; et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat. Cancer 2021, 2, 1071–1085. [Google Scholar] [CrossRef]
- Castro, A.; Zanetti, M.; Carter, H. Neoantigen Controversies. Annu. Rev. Biomed. Data Sci. 2021, 4, 227–253. [Google Scholar] [CrossRef]
- Black, J.R.M.; McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 2021, 21, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.W.; Yi, Z.; Weissman, J.S.; Chen, J. The dark proteome: Translation from noncanonical open reading frames. Trends Cell Biol. 2022, 32, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Starck, S.R.; Shastri, N. Nowhere to hide: Unconventional translation yields cryptic peptides for immune surveillance. Immunol. Rev. 2016, 272, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Rätsch, G. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e216. [Google Scholar] [CrossRef]
- Malarkannan, S.; Horng, T.; Shih, P.P.; Schwab, S.; Shastri, N. Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity 1999, 10, 681–690. [Google Scholar] [CrossRef]
- Schwab, S.R.; Shugart, J.A.; Horng, T.; Malarkannan, S.; Shastri, N. Unanticipated antigens: Translation initiation at CUG with leucine. PLoS Biol. 2004, 2, e366. [Google Scholar] [CrossRef]
- Jackson, R.; Kroehling, L.; Khitun, A.; Bailis, W.; Jarret, A.; York, A.G.; Khan, O.M.; Brewer, J.R.; Skadow, M.H.; Duizer, C.; et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature 2018, 564, 434–438. [Google Scholar] [CrossRef]
- Chong, C.; Coukos, G.; Bassani-Sternberg, M. Identification of tumor antigens with immunopeptidomics. Nat. Biotechnol. 2022, 40, 175–188. [Google Scholar] [CrossRef]
- Ouspenskaia, T.; Law, T.; Clauser, K.R.; Klaeger, S.; Sarkizova, S.; Aguet, F.; Li, B.; Christian, E.; Knisbacher, B.A.; Le, P.M.; et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 2022, 40, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, E.; Robinson, T.M.; Zook, M.B.; Flanigan, K.M.; Atkins, J.F.; Howard, M.T.; Eisenlohr, L.C. Cryptic MHC class I-binding peptides are revealed by aminoglycoside-induced stop codon read-through into the 3′ UTR. Proc. Natl. Acad. Sci. USA 2014, 111, 5670–5675. [Google Scholar] [CrossRef] [PubMed]
- Bullock, T.N.; Eisenlohr, L.C. Ribosomal scanning past the primary initiation codon as a mechanism for expression of CTL epitopes encoded in alternative reading frames. J. Exp. Med. 1996, 184, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Saulquin, X.; Scotet, E.; Trautmann, L.; Peyrat, M.A.; Halary, F.; Bonneville, M.; Houssaint, E. +1 Frameshifting as a novel mechanism to generate a cryptic cytotoxic T lymphocyte epitope derived from human interleukin 10. J. Exp. Med. 2002, 195, 353–358. [Google Scholar] [CrossRef]
- Malaker, S.A.; Penny, S.A.; Steadman, L.G.; Myers, P.T.; Loke, J.C.; Raghavan, M.; Bai, D.L.; Shabanowitz, J.; Hunt, D.F.; Cobbold, M. Identification of Glycopeptides as Posttranslationally Modified Neoantigens in Leukemia. Cancer Immunol. Res. 2017, 5, 376–384. [Google Scholar] [CrossRef]
- Vigneron, N.; Stroobant, V.; Ferrari, V.; Abi Habib, J.; Van den Eynde, B.J. Production of spliced peptides by the proteasome. Mol. Immunol. 2019, 113, 93–102. [Google Scholar] [CrossRef]
- Liepe, J.; Ovaa, H.; Mishto, M. Why do proteases mess up with antigen presentation by re-shuffling antigen sequences? Curr. Opin. Immunol. 2018, 52, 81–86. [Google Scholar] [CrossRef]
- Ruiz Cuevas, M.V.; Hardy, M.P.; Hollý, J.; Bonneil, É.; Durette, C.; Courcelles, M.; Lanoix, J.; Côté, C.; Staudt, L.M.; Lemieux, S.; et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021, 34, 108815. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi Najafabadi, S.A.; Bolhassani, A.; Aghasadeghi, M.R. Tumor cell-based vaccine: An effective strategy for eradication of cancer cells. Immunotherapy 2022, 14, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Berd, D. M-Vax: An autologous, hapten-modified vaccine for human cancer. Expert Opin. Biol. Ther. 2002, 2, 335–342. [Google Scholar] [CrossRef]
- Sosman, J.A.; Sondak, V.K. Melacine: An allogeneic melanoma tumor cell lysate vaccine. Expert Rev. Vaccines 2003, 2, 353–368. [Google Scholar] [CrossRef]
- Ogi, C.; Aruga, A. Clinical evaluation of therapeutic cancer vaccines. Hum. Vaccin. Immunother. 2013, 9, 1049–1057. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11, 27. [Google Scholar] [CrossRef]
- Bohnert, R.; Vivas, S.; Jansen, G. Comprehensive benchmarking of SNV callers for highly admixed tumor data. PLoS ONE 2017, 12, e0186175. [Google Scholar] [CrossRef]
- Pei, S.; Liu, T.; Ren, X.; Li, W.; Chen, C.; Xie, Z. Benchmarking variant callers in next-generation and third-generation sequencing analysis. Brief. Bioinform. 2021, 22, bbaa148. [Google Scholar] [CrossRef]
- Sarwal, V.; Niehus, S.; Ayyala, R.; Kim, M.; Sarkar, A.; Chang, S.; Lu, A.; Rajkumar, N.; Darfci-Maher, N.; Littman, R.; et al. A comprehensive benchmarking of WGS-based deletion structural variant callers. Brief. Bioinform. 2022, 23, bbac221. [Google Scholar] [CrossRef] [PubMed]
- Gallegos Ruiz, M.I.; Floor, K.; Rijmen, F.; Grünberg, K.; Rodriguez, J.A.; Giaccone, G. EGFR and K-ras mutation analysis in non-small cell lung cancer: Comparison of paraffin embedded versus frozen specimens. Cell Oncol. 2007, 29, 257–264. [Google Scholar] [CrossRef]
- Cazzato, G.; Caporusso, C.; Arezzo, F.; Cimmino, A.; Colagrande, A.; Loizzi, V.; Cormio, G.; Lettini, T.; Maiorano, E.; Scarcella, V.S.; et al. Formalin-Fixed and Paraffin-Embedded Samples for Next Generation Sequencing: Problems and Solutions. Genes 2021, 12, 1472. [Google Scholar] [CrossRef]
- Dodani, D.D.; Nguyen, M.H.; Morin, R.D.; Marra, M.A.; Corbett, R.D. Combinatorial and Machine Learning Approaches for Improved Somatic Variant Calling From Formalin-Fixed Paraffin-Embedded Genome Sequence Data. Front. Genet. 2022, 13, 834764. [Google Scholar] [CrossRef]
- Trevarton, A.J.; Chang, J.T.; Symmans, W.F. Simple combination of multiple somatic variant callers to increase accuracy. Sci. Rep. 2023, 13, 8463. [Google Scholar] [CrossRef]
- Junjun, R.; Zhengqian, Z.; Ying, W.; Jialiang, W.; Yongzhuang, L. A comprehensive review of deep learning-based variant calling methods. Brief. Funct. Genom. 2024, 23, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Quinn, E.M.; Cormican, P.; Kenny, E.M.; Hill, M.; Anney, R.; Gill, M.; Corvin, A.P.; Morris, D.W. Development of strategies for SNP detection in RNA-seq data: Application to lymphoblastoid cell lines and evaluation using 1000 Genomes data. PLoS ONE 2013, 8, e58815. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, K.; Wang, W.L.; Yin, T.T.; Dong, W.Q.; Xu, C.J. A high-throughput SNP discovery strategy for RNA-seq data. BMC Genom. 2019, 20, 160. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Y.; Zhang, L.; Li, Z.; Fang, Q.; Gao, R.; Zhang, Z. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019, 20, 242. [Google Scholar] [CrossRef]
- CPTAC. Pan-Cancer Analysis Page. National Cancer Institute Proteomic Data Commons. Available online: https://pdc.cancer.gov/pdc/ (accessed on 27 August 2025).
- Erhard, F.; Halenius, A.; Zimmermann, C.; L’Hernault, A.; Kowalewski, D.J.; Weekes, M.P.; Stevanovic, S.; Zimmer, R.; Dölken, L. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 2018, 15, 363–366. [Google Scholar] [CrossRef]
- Fedorova, A.D.; Tierney, J.A.S.; Michel, A.M.; Baranov, P.V. RiboGalaxy: A Galaxy-based Web Platform for Ribosome Profiling Data Processing—2023 Update. J. Mol. Biol. 2023, 435, 168043. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Kubiniok, P.; Kovalchik, K.A.; Ma, Q.; Duquette, J.D.; Mongrain, I.; Deutsch, E.W.; Peters, B.; Sette, A.; Sirois, I.; et al. The Human Immunopeptidome Project: A Roadmap to Predict and Treat Immune Diseases. Mol. Cell Proteom. 2020, 19, 31–49. [Google Scholar] [CrossRef]
- Di Marco, M.; Schuster, H.; Backert, L.; Ghosh, M.; Rammensee, H.G.; Stevanović, S. Unveiling the Peptide Motifs of HLA-C and HLA-G from Naturally Presented Peptides and Generation of Binding Prediction Matrices. J. Immunol. 2017, 199, 2639–2651. [Google Scholar] [CrossRef]
- Szolek, A.; Schubert, B.; Mohr, C.; Sturm, M.; Feldhahn, M.; Kohlbacher, O. OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics 2014, 30, 3310–3316. [Google Scholar] [CrossRef]
- Kiyotani, K.; Mai, T.H.; Nakamura, Y. Comparison of exome-based HLA class I genotyping tools: Identification of platform-specific genotyping errors. J. Hum. Genet. 2017, 62, 397–405. [Google Scholar] [CrossRef]
- Orenbuch, R.; Filip, I.; Comito, D.; Shaman, J.; Pe’er, I.; Rabadan, R. arcasHLA: High-resolution HLA typing from RNAseq. Bioinformatics 2020, 36, 33–40. [Google Scholar] [CrossRef]
- Liu, C.; Yang, X.; Duffy, B.; Mohanakumar, T.; Mitra, R.D.; Zody, M.C.; Pfeifer, J.D. ATHLATES: Accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res. 2013, 41, e142. [Google Scholar] [CrossRef]
- Boegel, S.; Löwer, M.; Schäfer, M.; Bukur, T.; de Graaf, J.; Boisguérin, V.; Türeci, O.; Diken, M.; Castle, J.C.; Sahin, U. HLA typing from RNA-Seq sequence reads. Genome Med. 2012, 4, 102. [Google Scholar] [CrossRef]
- Profaizer, T.; Lázár-Molnár, E.; Close, D.W.; Delgado, J.C.; Kumánovics, A. HLA genotyping in the clinical laboratory: Comparison of next-generation sequencing methods. Hla 2016, 88, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Røder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2007, 2, e796. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Karosiene, E.; Rasmussen, M.; Stryhn, A.; Buus, S.; Nielsen, M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015, 67, 641–650. [Google Scholar] [CrossRef]
- Gfeller, D.; Bassani-Sternberg, M. Predicting Antigen Presentation-What Could We Learn From a Million Peptides? Front. Immunol. 2018, 9, 1716. [Google Scholar] [CrossRef]
- IEDB Analysis Resource. Epitope Prediction and Analysis Tools. Available online: http://tools.iedb.org/main/ (accessed on 25 August 2025).
- Yang, Y.; Wei, Z.; Cia, G.; Song, X.; Pucci, F.; Rooman, M.; Xue, F.; Hou, Q. MHCII-peptide presentation: An assessment of the state-of-the-art prediction methods. Front. Immunol. 2024, 15, 1293706. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2018, 37, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Albert, B.A.; Yang, Y.; Shao, X.M.; Singh, D.; Smit, K.N.; Anagnostou, V.; Karchin, R. Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity. Nat. Mach. Intell. 2023, 5, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; Bagaev, D.V.; Zvyagin, I.V.; Vroomans, R.M.; Crawford, J.C.; Dolton, G.; Komech, E.A.; Sycheva, A.L.; Koneva, A.E.; Egorov, E.S.; et al. VDJdb: A curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018, 46, D419–D427. [Google Scholar] [CrossRef] [PubMed]
- Tickotsky, N.; Sagiv, T.; Prilusky, J.; Shifrut, E.; Friedman, N. McPAS-TCR: A manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 2017, 33, 2924–2929. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, L.; Liu, K.; Wei, X.; Yang, K.; Du, W.; Wang, S.; Guo, N.; Ma, C.; Luo, L.; et al. PIRD: Pan Immune Repertoire Database. Bioinformatics 2020, 36, 897–903. [Google Scholar] [CrossRef]
- Nolan, S.; Vignali, M.; Klinger, M.; Dines, J.N.; Kaplan, I.M.; Svejnoha, E.; Craft, T.; Boland, K.; Pesesky, M.; Gittelman, R.M.; et al. A large-scale database of T-cell receptor beta (TCRβ) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Res. Sq. 2020, 16, 1488851. [Google Scholar] [CrossRef]
- 10× Genomics. Chromium Single Cell Immune Profiling. Available online: https://www.10xgenomics.com/products/single-cell-immune-profiling (accessed on 8 August 2025).
- Fischer, D.S.; Wu, Y.; Schubert, B.; Theis, F.J. Predicting antigen specificity of single T cells based on TCR CDR3 regions. Mol. Syst. Biol. 2020, 16, e9416. [Google Scholar] [CrossRef]
- Overall, S.A.; Toor, J.S.; Hao, S.; Yarmarkovich, M.; Sara, M.O.R.; Morozov, G.I.; Nguyen, S.; Japp, A.S.; Gonzalez, N.; Moschidi, D.; et al. High throughput pMHC-I tetramer library production using chaperone-mediated peptide exchange. Nat. Commun. 2020, 11, 1909. [Google Scholar] [CrossRef]
- Meysman, P.; Barton, J.; Bravi, B.; Cohen-Lavi, L.; Karnaukhov, V.; Lilleskov, E.; Montemurro, A.; Nielsen, M.; Mora, T.; Pereira, P.; et al. Benchmarking solutions to the T-cell receptor epitope prediction problem: IMMREP22 workshop report. ImmunoInformatics 2023, 9, 100024. [Google Scholar] [CrossRef]
- Grazioli, F.; Mösch, A.; Machart, P.; Li, K.; Alqassem, I.; O’Donnell, T.J.; Min, M.R. On TCR binding predictors failing to generalize to unseen peptides. Front. Immunol. 2022, 13, 1014256. [Google Scholar] [CrossRef]
- Croce, G.; Bobisse, S.; Moreno, D.L.; Schmidt, J.; Guillame, P.; Harari, A.; Gfeller, D. Deep learning predictions of TCR-epitope interactions reveal epitope-specific chains in dual alpha T cells. Nat. Commun. 2024, 15, 3211. [Google Scholar] [CrossRef]
- Habern, O. 2021 A Sequencing Approach to T-Cell Receptor-Antigen Recognition. 10× Genomics. Available online: https://www.10xgenomics.com/blog/a-sequencing-approach-to-t-cell-receptor-antigen-recognition (accessed on 18 July 2025).
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef]
- Ragone, C.; Cavalluzzo, B.; Mauriello, A.; Tagliamonte, M.; Buonaguro, L. Lack of shared neoantigens in prevalent mutations in cancer. J. Transl. Med. 2024, 22, 344. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Bagley, S.J.; Binder, Z.A.; Lamrani, L.; Marinari, E.; Desai, A.S.; Nasrallah, M.P.; Maloney, E.; Brem, S.; Lustig, R.A.; Kurtz, G.; et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: A phase 1 trial. Nat. Cancer 2024, 5, 517–531. [Google Scholar] [CrossRef]
- Laganà, A. Computational Approaches for the Investigation of Intra-tumor Heterogeneity and Clonal Evolution from Bulk Sequencing Data in Precision Oncology Applications. Adv. Exp. Med. Biol. 2022, 1361, 101–118. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, X.; Zhang, H.; Ulintz, P.J.; Li, H.; Guan, Y. FastClone is a probabilistic tool for deconvoluting tumor heterogeneity in bulk-sequencing samples. Nat. Commun. 2020, 11, 4469. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Côté, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef]
- Miller, C.A.; White, B.S.; Dees, N.D.; Griffith, M.; Welch, J.S.; Griffith, O.L.; Vij, R.; Tomasson, M.H.; Graubert, T.A.; Walter, M.J.; et al. SciClone: Inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput. Biol. 2014, 10, e1003665. [Google Scholar] [CrossRef] [PubMed]
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.Y.; Graubert, A.P.; et al. pVACtools: A Computational Toolkit to Identify and Visualize Cancer Neoantigens. Cancer Immunol. Res. 2020, 8, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Kodysh, J.; Rubinsteyn, A. OpenVax: An Open-Source Computational Pipeline for Cancer Neoantigen Prediction. Methods Mol. Biol. 2020, 2120, 147–160. [Google Scholar] [CrossRef]
- Li, B.; Jing, P.; Zheng, G.; Pi, C.; Zhang, L.; Yin, Z.; Xu, L.; Qiu, J.; Gu, H.; Qiu, T.; et al. Neo-intline: Integrated pipeline enables neoantigen design through the in-silico presentation of T-cell epitope. Signal. Transduct. Target. Ther. 2023, 8, 397. [Google Scholar] [CrossRef]
- Rubinsteyn, A.; Kodysh, J.; Hodes, I.; Mondet, S.; Aksoy, B.A.; Finnigan, J.P.; Bhardwaj, N.; Hammerbacher, J. Computational Pipeline for the PGV-001 Neoantigen Vaccine Trial. Front. Immunol. 2017, 8, 1807. [Google Scholar] [CrossRef]
- Schmittel, A.; Keilholz, U.; Scheibenbogen, C. Evaluation of the interferon-gamma ELISPOT-assay for quantification of peptide specific T lymphocytes from peripheral blood. J. Immunol. Methods 1997, 210, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Godard, B.; Gazagne, A.; Gey, A.; Baptiste, M.; Vingert, B.; Pegaz-Fiornet, B.; Strompf, L.; Fridman, W.H.; Glotz, D.; Tartour, E. Optimization of an elispot assay to detect cytomegalovirus-specific CD8+ T lymphocytes. Hum. Immunol. 2004, 65, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Czerkinsky, C.C.; Nilsson, L.A.; Nygren, H.; Ouchterlony, O.; Tarkowski, A. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods 1983, 65, 109–121. [Google Scholar] [CrossRef]
- Slota, M.; Lim, J.B.; Dang, Y.; Disis, M.L. ELISpot for measuring human immune responses to vaccines. Expert Rev. Vaccines 2011, 10, 299–306. [Google Scholar] [CrossRef]
- Korzeniewski, C.; Callewaert, D.M. An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 1983, 64, 313–320. [Google Scholar] [CrossRef]
- Weigelin, B.; den Boer, A.T.; Wagena, E.; Broen, K.; Dolstra, H.; de Boer, R.J.; Figdor, C.G.; Textor, J.; Friedl, P. Cytotoxic T cells are able to efficiently eliminate cancer cells by additive cytotoxicity. Nat. Commun. 2021, 12, 5217. [Google Scholar] [CrossRef]
- Gao, S.; Wu, Z.; Arnold, B.; Diamond, C.; Batchu, S.; Giudice, V.; Alemu, L.; Raffo, D.Q.; Feng, X.; Kajigaya, S.; et al. Single-cell RNA sequencing coupled to TCR profiling of large granular lymphocyte leukemia T cells. Nat. Commun. 2022, 13, 1982. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.L.; Lu, K.; Erdogan, C.; Han, Y.; Hu, J.; Wang, T.; Heymach, J.V.; Zhang, J.; Reuben, A. T-Cell Receptor Repertoire Sequencing in the Era of Cancer Immunotherapy. Clin. Cancer Res. 2023, 29, 994–1008. [Google Scholar] [CrossRef]
- Kato, T.; Matsuda, T.; Ikeda, Y.; Park, J.H.; Leisegang, M.; Yoshimura, S.; Hikichi, T.; Harada, M.; Zewde, M.; Sato, S.; et al. Effective screening of T cells recognizing neoantigens and construction of T-cell receptor-engineered T cells. Oncotarget 2018, 9, 11009–11019. [Google Scholar] [CrossRef] [PubMed]
- Zong, S.; Mi, T.; Flores, L.G., 2nd; Alpert, A.; Olivares, S.; Patel, K.; Maiti, S.; McNamara, G.; Cooper, L.J.N.; Torikai, H. Very rapid cloning, expression and identifying specificity of T-cell receptors for T-cell engineering. PLoS ONE 2020, 15, e0228112. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef]
- Guil-Luna, S.; Sedlik, C.; Piaggio, E. Humanized Mouse Models to Evaluate Cancer Immunotherapeutics. Annu. Rev. Cancer Biol. 2021, 5, 119–136. [Google Scholar] [CrossRef]
- Zhang, X.; Kim, S.; Hundal, J.; Herndon, J.M.; Li, S.; Petti, A.A.; Soysal, S.D.; Li, L.; McLellan, M.D.; Hoog, J.; et al. Breast Cancer Neoantigens Can Induce CD8+ T-Cell Responses and Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 516–523. [Google Scholar] [CrossRef]
- Vijh, S.; Pilip, I.M.; Pamer, E.G. Effect of antigen-processing efficiency on in vivo T cell response magnitudes. J. Immunol. 1998, 160, 3971–3977. [Google Scholar] [CrossRef]
- Lopez, J.S.; Camidge, R.; Iafolla, M.; Rottey, S.; Schuler, M.; Hellmann, M.; Balmanoukian, A.; Dirix, L.; Gordon, M.; Sullivan, R.; et al. A phase Ib study to evaluate RO7198457, an individualized Neoantigen Specific immunoTherapy (iNeST), in combination with atezolizumab in patients with locally advanced or metastatic solid tumors. Cancer Res. 2020, 80, CT301. [Google Scholar] [CrossRef]
- Van Pul, K.M.; Fransen, M.F.; van de Ven, R.; de Gruijl, T.D. Immunotherapy Goes Local: The Central Role of Lymph Nodes in Driving Tumor Infiltration and Efficacy. Front. Immunol. 2021, 12, 643291. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Huang, R.R.; Lee, J.; Itakura, E.; Leong, S.P.; Essner, R. Tumour-induced immune modulation of sentinel lymph nodes. Nat. Rev. Immunol. 2006, 6, 659–670. [Google Scholar] [CrossRef]
- Norbury, C.C.; Basta, S.; Donohue, K.B.; Tscharke, D.C.; Princiotta, M.F.; Berglund, P.; Gibbs, J.; Bennink, J.R.; Yewdell, J.W. CD8+ T cell cross-priming via transfer of proteasome substrates. Science 2004, 304, 1318–1321. [Google Scholar] [CrossRef]
- Ho, N.I.; Huis In‘t Veld, L.G.M.; Raaijmakers, T.K.; Adema, G.J. Adjuvants Enhancing Cross-Presentation by Dendritic Cells: The Key to More Effective Vaccines? Front. Immunol. 2018, 9, 2874. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Middaugh, C.R. Barriers to nonviral gene delivery. J. Pharm. Sci. 2003, 92, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Hobernik, D.; Bros, M. DNA Vaccines-How Far From Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.N.; Guthrie, K.A.; Liu, Y.; Coveler, A.L.; Higgins, D.M.; Childs, J.S.; Dang, Y.; Salazar, L.G. Safety and Outcomes of a Plasmid DNA Vaccine Encoding the ERBB2 Intracellular Domain in Patients with Advanced-Stage ERBB2-Positive Breast Cancer: A Phase 1 Nonrandomized Clinical Trial. JAMA Oncol. 2023, 9, 71–78. [Google Scholar] [CrossRef]
- Schalk, J.A.; Mooi, F.R.; Berbers, G.A.; van Aerts, L.A.; Ovelgönne, H.; Kimman, T.G. Preclinical and clinical safety studies on DNA vaccines. Hum. Vaccin. 2006, 2, 45–53. [Google Scholar] [CrossRef]
- Fioretti, D.; Iurescia, S.; Rinaldi, M. Recent advances in design of immunogenic and effective naked DNA vaccines against cancer. Recent. Pat. Anticancer. Drug Discov. 2014, 9, 66–82. [Google Scholar] [CrossRef]
- Faurez, F.; Dory, D.; Le Moigne, V.; Gravier, R.; Jestin, A. Biosafety of DNA vaccines: New generation of DNA vectors and current knowledge on the fate of plasmids after injection. Vaccine 2010, 28, 3888–3895. [Google Scholar] [CrossRef] [PubMed]
- Khobragade, A.; Bhate, S.; Ramaiah, V.; Deshpande, S.; Giri, K.; Phophle, H.; Supe, P.; Godara, I.; Revanna, R.; Nagarkar, R.; et al. Efficacy, safety, and immunogenicity of the DNA SARS-CoV-2 vaccine (ZyCoV-D): The interim efficacy results of a phase 3, randomised, double-blind, placebo-controlled study in India. Lancet 2022, 399, 1313–1321. [Google Scholar] [CrossRef]
- Ndeupen, S.; Qin, Z.; Jacobsen, S.; Bouteau, A.; Estanbouli, H.; Igyártó, B.Z. The mRNA-LNP platform’s lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience 2021, 24, 103479. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Liu, M.A. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef]
- Ledwith, B.J.; Manam, S.; Troilo, P.J.; Barnum, A.B.; Pauley, C.J.; Griffiths, T.G., 2nd; Harper, L.B.; Beare, C.M.; Bagdon, W.J.; Nichols, W.W. Plasmid DNA vaccines: Investigation of integration into host cellular DNA following intramuscular injection in mice. Intervirology 2000, 43, 258–272. [Google Scholar] [CrossRef]
- Li, Y.D.; Chi, W.Y.; Su, J.H.; Ferrall, L.; Hung, C.F.; Wu, T.C. Coronavirus vaccine development: From SARS and MERS to COVID-19. J. Biomed. Sci. 2020, 27, 104. [Google Scholar] [CrossRef] [PubMed]
- Soltani, S.; Farahani, A.; Dastranj, M.; Momenifar, N.; Mohajeri, P.; Emamie, A.D. Dna Vaccine: Methods and Mechanisms. Adv. Hum. Biol. 2018, 8, 132–139. [Google Scholar] [CrossRef]
- Jorritsma, S.H.T.; Gowans, E.J.; Grubor-Bauk, B.; Wijesundara, D.K. Delivery methods to increase cellular uptake and immunogenicity of DNA vaccines. Vaccine 2016, 34, 5488–5494. [Google Scholar] [CrossRef]
- Hauser, H.; Chen, S.Y. Augmentation of DNA vaccine potency through secretory heat shock protein-mediated antigen targeting. Methods 2003, 31, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Trimble, C.L.; Peng, S.; Kos, F.; Gravitt, P.; Viscidi, R.; Sugar, E.; Pardoll, D.; Wu, T.C. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin. Cancer Res. 2009, 15, 361–367. [Google Scholar] [CrossRef]
- Hung, C.F.; Hsu, K.F.; Cheng, W.F.; Chai, C.Y.; He, L.; Ling, M.; Wu, T.C. Enhancement of DNA vaccine potency by linkage of antigen gene to a gene encoding the extracellular domain of Fms-like tyrosine kinase 3-ligand. Cancer Res. 2001, 61, 1080–1088. [Google Scholar]
- Choi, Y.J.; Hur, S.Y.; Kim, T.J.; Hong, S.R.; Lee, J.K.; Cho, C.H.; Park, K.S.; Woo, J.W.; Sung, Y.C.; Suh, Y.S.; et al. A Phase II, Prospective, Randomized, Multicenter, Open-Label Study of GX-188E, an HPV DNA Vaccine, in Patients with Cervical Intraepithelial Neoplasia 3. Clin. Cancer Res. 2020, 26, 1616–1623. [Google Scholar] [CrossRef]
- Cheng, W.F.; Hung, C.F.; Chai, C.Y.; Hsu, K.F.; He, L.; Ling, M.; Wu, T.C. Tumor-specific immunity and antiangiogenesis generated by a DNA vaccine encoding calreticulin linked to a tumor antigen. J. Clin. Investig. 2001, 108, 669–678. [Google Scholar] [CrossRef]
- Garcia, F.; Petry, K.U.; Muderspach, L.; Gold, M.A.; Braly, P.; Crum, C.P.; Magill, M.; Silverman, M.; Urban, R.G.; Hedley, M.L.; et al. ZYC101a for treatment of high-grade cervical intraepithelial neoplasia: A randomized controlled trial. Obstet. Gynecol. 2004, 103, 317–326. [Google Scholar] [CrossRef]
- Matijevic, M.; Hedley, M.L.; Urban, R.G.; Chicz, R.M.; Lajoie, C.; Luby, T.M. Immunization with a poly (lactide co-glycolide) encapsulated plasmid DNA expressing antigenic regions of HPV 16 and 18 results in an increase in the precursor frequency of T cells that respond to epitopes from HPV 16, 18, 6 and 11. Cell Immunol. 2011, 270, 62–69. [Google Scholar] [CrossRef]
- Kabachinski, G.; Schwartz, T.U. The nuclear pore complex--structure and function at a glance. J. Cell Sci. 2015, 128, 423–429. [Google Scholar] [CrossRef]
- Dean, D.A. Cell-specific targeting strategies for electroporation-mediated gene delivery in cells and animals. J. Membr. Biol. 2013, 246, 737–744. [Google Scholar] [CrossRef]
- Cervia, L.D.; Yuan, F. Current Progress in Electrotransfection as a Nonviral Method for Gene Delivery. Mol. Pharm. 2018, 15, 3617–3624. [Google Scholar] [CrossRef]
- Dietz, W.M.; Skinner, N.E.; Hamilton, S.E.; Jund, M.D.; Heitfeld, S.M.; Litterman, A.J.; Hwu, P.; Chen, Z.Y.; Salazar, A.M.; Ohlfest, J.R.; et al. Minicircle DNA is superior to plasmid DNA in eliciting antigen-specific CD8+ T-cell responses. Mol. Ther. 2013, 21, 1526–1535. [Google Scholar] [CrossRef]
- Chen, Z.Y.; He, C.Y.; Ehrhardt, A.; Kay, M.A. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol. Ther. 2003, 8, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Boye, C.; Arpag, S.; Francis, M.; DeClemente, S.; West, A.; Heller, R.; Bulysheva, A. Reduction of plasmid vector backbone length enhances reporter gene expression. Bioelectrochemistry 2022, 144, 107981. [Google Scholar] [CrossRef] [PubMed]
- Darquet, A.M.; Cameron, B.; Wils, P.; Scherman, D.; Crouzet, J. A new DNA vehicle for nonviral gene delivery: Supercoiled minicircle. Gene Ther. 1997, 4, 1341–1349. [Google Scholar] [CrossRef]
- Bigger, B.W.; Tolmachov, O.; Collombet, J.M.; Fragkos, M.; Palaszewski, I.; Coutelle, C. An araC-controlled bacterial cre expression system to produce DNA minicircle vectors for nuclear and mitochondrial gene therapy. J. Biol. Chem. 2001, 276, 23018–23027. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, P.; Blaesen, M.; Schleef, M.; Jechlinger, W. Minicircle-DNA production by site specific recombination and protein-DNA interaction chromatography. J. Gene Med. 2008, 10, 1253–1269. [Google Scholar] [CrossRef]
- Williams, J.A.; Luke, J.; Johnson, L.; Hodgson, C. pDNAVACCultra vector family: High throughput intracellular targeting DNA vaccine plasmids. Vaccine 2006, 24, 4671–4676. [Google Scholar] [CrossRef]
- Luke, J.; Carnes, A.E.; Hodgson, C.P.; Williams, J.A. Improved antibiotic-free DNA vaccine vectors utilizing a novel RNA based plasmid selection system. Vaccine 2009, 27, 6454–6459. [Google Scholar] [CrossRef]
- Williams, J.A.; Paez, P.A. Improving cell and gene therapy safety and performance using next-generation Nanoplasmid vectors. Mol. Ther. Nucleic Acids 2023, 32, 494–503. [Google Scholar] [CrossRef]
- Adie, T.; Orefo, I.; Kysh, D.; Kondas, K.; Thapa, S.; Extance, J.; Duncan, T.; Rothwell, P.J. dbDNA™: An advanced platform for genetic medicines. Drug Discov. Today 2022, 27, 374–377. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, D.; McLellan, M.; Zhang, X.; Vickery, T.; Myers, N.; Sturmoski, M.; Ruzinova, M.; Hundal, J.; Miller, C.; Griffith, M.; et al. Preliminary Results Of A Phase Ib Clinical Trial Of A Neoantigen Dna Vaccine For Pancreatic Cancer. HPB 2020, 22, S12–S13. [Google Scholar] [CrossRef]
- Ochsenreither, S.; Anguera, G.; Dieter, S.; Trikalinos, N.; Fu, S.; Fernández, B.C.; Ferrándiz, A.C.; Kim, S.Y.; Rodriguez, L.M.; Berg, K.C.G.; et al. Induction of neoantigen-specific immune responses by VB10.NEO in combination with atezolizumab in heavily pretreated patients with advanced solid tumors: Final analysis of the phase 1b VB N-02 trial. J. Clin. Oncol. 2025, 43, 2639. [Google Scholar] [CrossRef]
- Shah, K.; Ganapathy, A.; Borkowski, A.; Shah, N.; Bansal, D.; Beck, R.; Knoche, E.M.; Picus, J.; Reimers, M.A.; Roth, B.J.; et al. A pilot trial of neoantigen DNA vaccine in combination with nivolumab/ipilimumab and prostvac in metastatic hormone-sensitive prostate cancer (mHSPC). J. Clin. Oncol. 2022, 40, 5068. [Google Scholar] [CrossRef]
- Johanns, T.M.; Garfinkle, E.A.R.; Miller, K.E.; Livingstone, A.J.; Roberts, K.F.; Rao Venkata, L.P.; Dowling, J.L.; Chicoine, M.R.; Dacey, R.G.; Zipfel, G.J.; et al. Integrating Multisector Molecular Characterization into Personalized Peptide Vaccine Design for Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2024, 30, 2729–2742. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Neoantigen-based Personalized DNA Vaccine with Retifanlimab PD-1 Blockade Therapy in Patients with Newly Diagnosed, Unmethylated Glioblastoma. (NCT05743595). Identifier: NCT05743595. Available online: https://clinicaltrials.gov/study/NCT05743595 (accessed on 27 August 2025).
- ClinicalTrials.gov. Neoepitope_based Personalized DNA Vaccine Approach in Pediatric Patients with Recurrent Brain Tumors. (NCT03988283). Identifier: NCT03988283. Available online: https://clinicaltrials.gov/study/NCT03988283 (accessed on 27 August 2025).
- ClinicalTrials.gov. PolyImmune {Durvalumad (MEDI4736) and Tremelimumab} & Vaccine Ochaestrated Treatment for Patients with Advanced/Metastatic Renal Cell Carcinoma (PIVOT-RCC). (NCT03598816). Identifier: NCT03598816. Available online: https://clinicaltrials.gov/study/NCT03598816 (accessed on 27 August 2025).
- ClinicalTrials.gov. Personalized Neoantigen Vaccine in Combination with Durvalumab (MEDI4736) in Extensive Stage Small Cell Lung Cancer. (NCT04397003) Identifier: NCT04397003. Available online: https://clinicaltrials.gov/study/NCT04397003 (accessed on 27 August 2025).
- Daniela, K.-K.; Nadia, V.; Michail, A.P.; Mads, L.; Thomas, T.; Nikolas, H.T.; Christian, G.; Anders, J.; Thomas, S.J.; Britt, W.L.; et al. 623 AI-designed personalized neoantigen vaccine, EVX-02, induces robust T-cell responses in melanoma patients. J. Immunother. Cancer 2023, 11, 623. [Google Scholar] [CrossRef]
- Thomas, S.K.; Cha, S.C.; Smith, D.L.; Kim, K.H.; Parshottam, S.R.; Rao, S.; Popescu, M.; Lee, V.Y.; Neelapu, S.S.; Kwak, L.W. Phase I study of an active immunotherapy for asymptomatic phase Lymphoplasmacytic lymphoma with DNA vaccines encoding antigen-chemokine fusion: Study protocol. BMC Cancer 2018, 18, 187. [Google Scholar] [CrossRef] [PubMed]
- Szymura, S.J.; Wang, L.; Zhang, T.; Cha, S.C.; Song, J.; Dong, Z.; Anderson, A.; Oh, E.; Lee, V.; Wang, Z.; et al. Personalized neoantigen vaccines as early intervention in untreated patients with lymphoplasmacytic lymphoma: A non-randomized phase 1 trial. Nat. Commun. 2024, 15, 6874. [Google Scholar] [CrossRef] [PubMed]
- King, D.A.; Smith, A.R.; Pineda, G.; Nakano, M.; Michelini, F.; Goedegebuure, S.P.; Thyparambil, S.; Liao, W.-L.; McCormick, A.; Ju, J.; et al. Complete Remission of Widely Metastatic Human Epidermal Growth Factor Receptor 2–Amplified Pancreatic Adenocarcinoma After Precision Immune and Targeted Therapy with Description of Sequencing and Organoid Correlates. JCO Precis. Oncol. 2023, 7, e2100489. [Google Scholar] [CrossRef]
- Garfinkle, E.A.; Miller, K.E.; Livingstone, A.J.; Perales-Linares, R.; Cooch, N.; Perales-Puchalt, A.; Rochestie, S.; Peters, J.; Sardesai, N.Y.; Gillanders, W.E.; et al. Abstract 1174: Incorporation of multisector analysis into the design of personalized DNA vaccines for patients with newly diagnosed glioblastoma. Cancer Res. 2024, 84, 1174. [Google Scholar] [CrossRef]
- Pandya, A.; Shah, Y.; Kothari, N.; Postwala, H.; Shah, A.; Parekh, P.; Chorawala, M.R. The future of cancer immunotherapy: DNA vaccines leading the way. Med. Oncol. 2023, 40, 200. [Google Scholar] [CrossRef]
- Xia, Q.; Zhang, F.F.; Geng, F.; Liu, C.L.; Wang, Y.Q.; Xu, P.; Lu, Z.Z.; Xie, Y.; Wu, H.; Chen, Y.; et al. Improvement of anti-tumor immunity of fibroblast activation protein α based vaccines by combination with cyclophosphamide in a murine model of breast cancer. Cell Immunol. 2016, 310, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jagodinsky, J.C.; Harari, P.M.; Morris, Z.S. The Promise of Combining Radiation Therapy with Immunotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 6–16. [Google Scholar] [CrossRef]
- Rupp, T.; Genest, L.; Babin, D.; Legrand, C.; Hunault, M.; Froget, G.; Castagné, V. Anti-CTLA-4 and anti-PD-1 immunotherapies repress tumor progression in preclinical breast and colon model with independent regulatory T cells response. Transl. Oncol. 2022, 20, 101405. [Google Scholar] [CrossRef]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 blockade drives loss of T(reg) stability in glycolysis-low tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef]
- Soong, R.S.; Trieu, J.; Lee, S.Y.; He, L.; Tsai, Y.C.; Wu, T.C.; Hung, C.F. Xenogeneic human p53 DNA vaccination by electroporation breaks immune tolerance to control murine tumors expressing mouse p53. PLoS ONE 2013, 8, e56912. [Google Scholar] [CrossRef]
- Yang, B.; Jeang, J.; Yang, A.; Wu, T.C.; Hung, C.F. DNA vaccine for cancer immunotherapy. Hum. Vaccin. Immunother. 2014, 10, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Propper, D.J.; Chao, D.; Braybrooke, J.P.; Bahl, P.; Thavasu, P.; Balkwill, F.; Turley, H.; Dobbs, N.; Gatter, K.; Talbot, D.C.; et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin. Cancer Res. 2003, 9, 84–92. [Google Scholar]
- Parikh, F.; Duluc, D.; Imai, N.; Clark, A.; Misiukiewicz, K.; Bonomi, M.; Gupta, V.; Patsias, A.; Parides, M.; Demicco, E.G.; et al. Chemoradiotherapy-induced upregulation of PD-1 antagonizes immunity to HPV-related oropharyngeal cancer. Cancer Res. 2014, 74, 7205–7216. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- van Meir, H.; Nout, R.A.; Welters, M.J.; Loof, N.M.; de Kam, M.L.; van Ham, J.J.; Samuels, S.; Kenter, G.G.; Cohen, A.F.; Melief, C.J.; et al. Impact of (chemo)radiotherapy on immune cell composition and function in cervical cancer patients. Oncoimmunology 2017, 6, e1267095. [Google Scholar] [CrossRef] [PubMed]
- Dersh, D.; Phelan, J.D.; Gumina, M.E.; Wang, B.; Arbuckle, J.H.; Holly, J.; Kishton, R.J.; Markowitz, T.E.; Seedhom, M.O.; Fridlyand, N.; et al. Genome-wide Screens Identify Lineage- and Tumor-Specific Genes Modulating MHC-I- and MHC-II-Restricted Immunosurveillance of Human Lymphomas. Immunity 2021, 54, 116–131.e110. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.; Pich, O.; Thol, K.; Watkins, T.B.K.; Luebeck, J.; Rowan, A.; Stavrou, G.; Weiser, N.E.; Dameracharla, B.; Bentham, R.; et al. Origins and impact of extrachromosomal DNA. Nature 2024, 635, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Pecorino, L.T.; Verhaak, R.G.W.; Henssen, A.; Mischel, P.S. Extrachromosomal DNA (ecDNA): An origin of tumor heterogeneity, genomic remodeling, and drug resistance. Biochem. Soc. Trans. 2022, 50, 1911–1920. [Google Scholar] [CrossRef]
- Bafna, V.; Mischel, P.S. Extrachromosomal DNA in Cancer. Annu. Rev. Genom. Hum. Genet. 2022, 23, 29–52. [Google Scholar] [CrossRef]
- Melief, C.J.; van der Burg, S.H. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat. Rev. Cancer 2008, 8, 351–360. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, J.; Lee, H.; Batista, M.T.; Johnston, S.A. RNA Transcription and Splicing Errors as a Source of Cancer Frameshift Neoantigens for Vaccines. Sci. Rep. 2019, 9, 14184. [Google Scholar] [CrossRef]
- Lange, J.T.; Rose, J.C.; Chen, C.Y.; Pichugin, Y.; Xie, L.; Tang, J.; Hung, K.L.; Yost, K.E.; Shi, Q.; Erb, M.L.; et al. The evolutionary dynamics of extrachromosomal DNA in human cancers. Nat. Genet. 2022, 54, 1527–1533. [Google Scholar] [CrossRef]
- Wu, A.A.; Drake, V.; Huang, H.-S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. OncoImmunology 2015, 4, e1016700. [Google Scholar] [CrossRef]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Iwadate, D.; Kato, H.; Nakai, Y.; Tateishi, K.; Fujishiro, M. Targeting the Metabolic Rewiring in Pancreatic Cancer and Its Tumor Microenvironment. Cancers 2022, 14, 4351. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Lee, J.H.; Massagué, J. TGF-β in developmental and fibrogenic EMTs. Semin. Cancer Biol. 2022, 86, 136–145. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7, re8. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, K.; Soares, L.; Engleman, E.G.; Merad, M. Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J. Clin. Investig. 2004, 113, 774–783. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Finn, O.J. The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 2018, 18, 183–194. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef]
- Melief, C.J. Cancer immunotherapy by dendritic cells. Immunity 2008, 29, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Gabai, Y.; Assouline, B.; Ben-Porath, I. Senescent stromal cells: Roles in the tumor microenvironment. Trends Cancer 2023, 9, 28–41. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef]
- Saraiva, M.; Vieira, P.; O’Garra, A. Biology and therapeutic potential of interleukin-10. J. Exp. Med. 2020, 217, e20190418. [Google Scholar] [CrossRef]
- Farmer, E.; Cheng, M.A.; Hung, C.F.; Wu, T.C. Vaccination Strategies for the Control and Treatment of HPV Infection and HPV-Associated Cancer. Recent Results Cancer Res. 2021, 217, 157–195. [Google Scholar] [CrossRef]
- Hauge, A.; Rofstad, E.K. Antifibrotic therapy to normalize the tumor microenvironment. J. Transl. Med. 2020, 18, 207. [Google Scholar] [CrossRef]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Lee, N.Y.; Sherman, E.J.; Schöder, H.; Wray, R.; Boyle, J.O.; Singh, B.; Grkovski, M.; Paudyal, R.; Cunningham, L.; Zhang, Z.; et al. Hypoxia-Directed Treatment of Human Papillomavirus-Related Oropharyngeal Carcinoma. J. Clin. Oncol. 2024, 42, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Powell, J.D. Fueling the Revolution: Targeting Metabolism to Enhance Immunotherapy. Cancer Immunol. Res. 2021, 9, 255–260. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Douek, D.C. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harb. Perspect. Med. 2011, 1, a007252. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Organ System | Vaccine Name | Vaccine Target | Target Disease | Delivery Method | Combination | Phase | Status | Outcome | NCT Number |
|---|---|---|---|---|---|---|---|---|---|
| Lymphatic System | scFv-CCL20 plasmid DNA vaccine | MIP3α-fused lymphoma idiotype | Lymphoplasmacytic lymphoma | DNA plasmid via I.D. injection with a needle-free bioinjector | I | Active, not recruiting | No results posted | NCT01209871 [203,204] | |
| Breast | Personalized polyepitope DNA vaccine | 4–20 patient specific neoantigens | TNBC | DNA plasmid via I.M. injection and electroporation | - | I | Complete | 36-month recurrence-free survival: 87.5% | NCT02348320 [4] |
| Pancreas | Neoantigen DNA vaccine | Personalized neoantigen DNA vaccine | Pancreatic Cancer—surgically resected with adjuvant chemotherapy without evidence of recurrent disease | DNA plasmid via I.M. injection and electroporation | Chemotherapy | I | Terminated due to loss of funding | Treatment-related adverse events grade ≤ 3: 7/7 | NCT03122106 [203] |
| Skin, Lung, Kidney, Bladder, Head and Neck, Breast, Cervix, Anus, Stomach/Esophagus, Colon | VB10.NEO | Up to 40 patient-specific neoantigens | Solid tumors | DNA plasmid via I.M. injection with a needle-free bioinjector | Atezolizumab—CPI (anti-PD-1 or anti-PD-L1), Bempegaldesleukin | I II/III | Complete | Stable disease: 34.8% (8/23) Neoantigen-specific response in stable disease patients: 100% (8/8) | NCT03548467 NCT05018273 [204] |
| Prostate | Neoantigen DNA vaccine | Patient-specific neoantigens | Prostate cancer | DNA plasmid via I.M. injection and electroporation | Nivolumab (anti-PD-1), Ipilimumab (anti-CTLA-4), PROSTVAC (PSA vaccine) | I | Complete | Treatment-related adverse events: Grade 3—13.3% (2/15) Grade ≤ 2: 86.7% (13/15) | NCT03532217 [205] |
| Liver | GNOS-PV02 | Up to 40 patient-specific neoantigens | Hepatocellular carcinoma | DNA plasmid via I.D. injection and electroporation | INO-9012 and pembrolizumab | I/II | Active, not recruiting | Objective response: 30.6% (11/36) Complete response: 8.3% (3/36) | NCT04251117 [5] |
| Brain/Meninges | GNOS-PV01 | Up to 30 antigens (27 TSA and 3 patient-specific) | Unmethylated glioblastoma | DNA plasmid via I.M. injection and electroporation | INO-9012 (plasmid encoding IL-12) | I | Active, not recruiting | No results posted | NCT04015700 [206] |
| Personalized neoantigen DNA vaccine | Patient-specific neoantigens | Glioblastoma | DNA plasmid via I.M. injection and electroporation | Retifanlimab | I | Recruiting | No results posted | NCT05743595 [207] | |
| Personalized neoantigen DNA vaccine | Patient-specific neoantigens | Brain tumors | DNA plasmid via I.M. injection and electroporation | - | I | Recruiting | No results posted | NCT03988283 [208] | |
| Kidney | Neoantigen DNA vaccine | Personalized neoantigen | Metastatic/advanced (inoperable) RCC | DNA plasmid via I.M. injection and electroporation | Durvalumab and tremelimumab | II | Withdrawn, FDA contingencies unresolved | No results posted | NCT03598816 [209] |
| Lungs | Neoantigen DNA vaccine | Patient-specific neoantigens | SCLC | DNA plasmid via I.M. injection and electroporation | Durvalumab, (and Carboplatin, Etoposide) | II | Active, not recruiting | No results posted | NCT04397003 [210] |
| Skin | EVX-02 | 13 neoantigens | Melanoma | I.M. injection | Nivolumab (anti-PD-1) | I/IIa | Terminated | No results posted | NCT04455503 [211] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, A.A.; Peng, K.; Vukovich, M.; Zhu, M.; Lin, Y.; Bagga, A.; Wu, T.; Hung, C.-F. Personalizing DNA Cancer Vaccines. J. Pers. Med. 2025, 15, 474. https://doi.org/10.3390/jpm15100474
Wu AA, Peng K, Vukovich M, Zhu M, Lin Y, Bagga A, Wu T, Hung C-F. Personalizing DNA Cancer Vaccines. Journal of Personalized Medicine. 2025; 15(10):474. https://doi.org/10.3390/jpm15100474
Chicago/Turabian StyleWu, Annie A., Kaiqi Peng, Melanie Vukovich, Michelle Zhu, Yuki Lin, Arindam Bagga, TC Wu, and Chien-Fu Hung. 2025. "Personalizing DNA Cancer Vaccines" Journal of Personalized Medicine 15, no. 10: 474. https://doi.org/10.3390/jpm15100474
APA StyleWu, A. A., Peng, K., Vukovich, M., Zhu, M., Lin, Y., Bagga, A., Wu, T., & Hung, C.-F. (2025). Personalizing DNA Cancer Vaccines. Journal of Personalized Medicine, 15(10), 474. https://doi.org/10.3390/jpm15100474