
Sudden Cardiac Death, Post-Mortem Investigation: A Proposing Panel of First Line and Second Line Genetic Tests

 , , , ,
, , , ,  , and
, and 
Abstract
1. Introduction
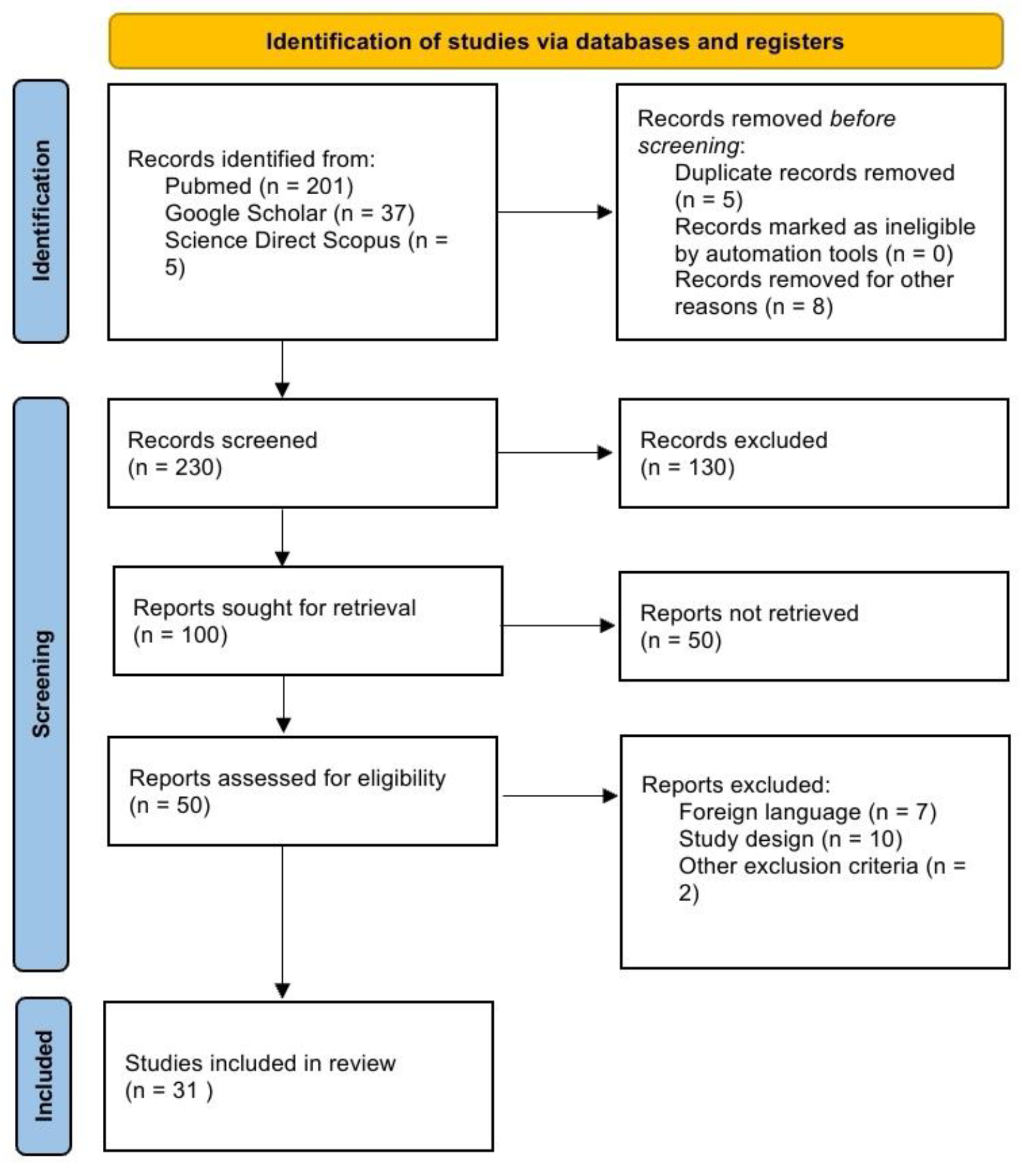
2. Materials and Methods
3. Results
3.1. Cardiomyophaty
3.2. Channelopathy and Arrhythmic Condition
4. Discussion
4.1. Hypertrophic Cardiomyopathy
4.2. Dilated Cardiomyopathy
4.3. Arrhythmogenic Cardiomyopathy
4.4. Channelopathy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fineschi, V.; Baroldi, G.; Silver, M.D. Pathology of the Heart and Sudden Death in Forensic Medicine; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar] [CrossRef]
- Payne-James, J.; Byard, R. Forensic and Legal Medicine: Clinical and Pathological Aspects, 1st ed.; CRC Press: Boca Raton, FL, USA, 2023. [Google Scholar] [CrossRef]
- Chugh, S.S.; Reinier, K.; Teodorescu, C.; Evanado, A.; Kehr, E.; Al Samara, M.; Mariani, R.; Gunson, K.; Jui, J. Epidemiology of Sudden Cardiac Death: Clinical and Research Implications. Prog. Cardiovasc. Dis. 2008, 51, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Yow, A.G.; Rajasurya, V.; Sharma, S. Sudden Cardiac Death. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK507854/ (accessed on 28 March 2024).
- Bailey, D.D. The Royal Collage of Pathologist, Guidelines on Autopsy Practice: Sudden Death with Likely Cardiac Pathology. Available online: https://www.ihrdni.org/314-008-1.pdf (accessed on 28 March 2024).
- Papadakis, M.; Papatheodorou, E.; Mellor, G.; Raju, H.; Bastiaenen, R.; Wijeyeratne, Y.; Wasim, S.; Ensam, B.; Finocchiaro, G.; Gray, B.; et al. The Diagnostic Yield of Brugada Syndrome After Sudden Death With Normal Autopsy. J. Am. Coll. Cardiol. 2018, 71, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Will, M.L.; Atkins, D.L.; Ackerman, M.J. A novel TPM1 mutation in a family with hypertrophic cardiomyopathy and sudden cardiac death in childhood. Am. J. Cardiol. 2002, 90, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Tiziana Di Gioia, C.R.; Autore, C.; Romeo, D.M.; Ciallella, C.; Aromatario, M.R.; Lopez, A.; Pagannone, E.; Giordano, C.; Gallo, P.; d’Amati, G. Sudden cardiac death in younger adults: Autopsy diagnosis as a tool for preventive medicine. Hum. Pathol. 2006, 37, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.K.; Nissen, P.H.; Berge, K.E.; Leren, T.P.; Kristensen, I.B.; Jensen, H.K.; Banner, J. Molecular autopsy in young sudden cardiac death victims with suspected cardiomyopathy. Forensic Sci. Int. 2012, 219, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sanchez-Molero, O.; Allegue, C.; Coll, M.; Mademont-Soler, I.; Selga, E.; Ferrer-Costa, C.; Mates, J.; Iglesias, A.; Sarquella-Brugada, G.; et al. Post-mortem genetic analysis in juvenile cases of sudden cardiac death. Forensic Sci. Int. 2014, 245, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Santori, M.; Blanco-Verea, A.; Gil, R.; Cortis, J.; Becker, K.; Schneider, P.M.; Carracedo, A.; Brion, M. Broad-based molecular autopsy: A potential tool to investigate the involvement of subtle cardiac conditions in sudden unexpected death in infancy and early childhood. Arch. Dis. Child. 2015, 100, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Tester, D.J.; Paulmichl, A.; Maleszewski, J.J.; Ackerman, M.J. Post-mortem Whole Exome Sequencing with Gene-Specific Analysis for Autopsy-Negative Sudden Unexplained Death in the Young: A Case Series. Pediatr. Cardiol. 2015, 36, 768–778. [Google Scholar] [CrossRef]
- LuCamp; Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-generation sequencing of 100 candidate genes in young victims of suspected sudden cardiac death with structural abnormalities of the heart. Int. J. Leg. Med. 2016, 130, 91–102. [Google Scholar] [CrossRef]
- Neubauer, J.; Haas, C.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W. Post-mortem whole-exome sequencing (WES) with a focus on cardiac disease-associated genes in five young sudden unexplained death (SUD) cases. Int. J. Leg. Med. 2016, 130, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Hellenthal, N.; Gaertner-Rommel, A.; Klauke, B.; Paluszkiewicz, L.; Stuhr, M.; Kerner, T.; Farr, M.; Püschel, K.; Milting, H. Molecular autopsy of sudden unexplained deaths reveals genetic predispositions for cardiac diseases among young forensic cases. Europace 2017, 19, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sanchez-Molero, O.; Fernandez, A.; Mademont-Soler, I.; Coll, M.; Perez-Serra, A.; Mates, J.; Del Olmo, B.; Pico, F.; Nogue-Navarro, L.; et al. Sudden Arrhythmic Death During Exercise: A Post-Mortem Genetic Analysis. Sports Med. 2017, 47, 2101–2115. [Google Scholar] [CrossRef]
- Andersen, J.D.; Jacobsen, S.B.; Trudsø, L.C.; Kampmann, M.-L.; Banner, J.; Morling, N. Whole genome and transcriptome sequencing of post-mortem cardiac tissues from sudden cardiac death victims identifies a gene regulatory variant in NEXN. Int. J. Leg. Med. 2019, 133, 1699–1709. [Google Scholar] [CrossRef]
- Marey, I.; Fressart, V.; Rambaud, C.; Fornes, P.; Martin, L.; Grotto, S.; Alembik, Y.; Gorka, H.; Millat, G.; Gandjbakhch, E.; et al. Clinical impact of post-mortem genetic testing in cardiac death and cardiomyopathy. Open Med. 2020, 15, 435–446. [Google Scholar] [CrossRef]
- Grassi, S.; Campuzano, O.; Coll, M.; Brión, M.; Arena, V.; Iglesias, A.; Carracedo, Á.; Brugada, R.; Oliva, A. Genetic variants of uncertain significance: How to match scientific rigour and standard of proof in sudden cardiac death? Leg. Med. 2020, 45, 101712. [Google Scholar] [CrossRef] [PubMed]
- Fernlund, E.; Kissopoulou, A.; Green, H.; Karlsson, J.-E.; Ellegård, R.; Årstrand, H.K.; Jonasson, J.; Gunnarsson, C. Hereditary Hypertrophic Cardiomyopathy in Children and Young Adults—The Value of Reevaluating and Expanding Gene Panel Analyses. Genes 2020, 11, 1472. [Google Scholar] [CrossRef]
- Fahed, A.C.; Nemer, G.; Bitar, F.F.; Arnaout, S.; Abchee, A.B.; Batrawi, M.; Khalil, A.; Abou Hassan, O.K.; DePalma, S.R.; McDonough, B.; et al. Founder Mutation in N Terminus of Cardiac Troponin I Causes Malignant Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 444–452. [Google Scholar] [CrossRef]
- Webster, G.; Reynolds, M.; Arva, N.C.; Dellefave-Castillo, L.M.; McElligott, H.S.; Kofman, A.; Laboski, A.; Magnetta, D.; George, A.L.; McNally, E.M.; et al. Mitochondrial cardiomyopathy and ventricular arrhythmias associated with biallelic variants in C1QBP. Am. J. Med. Genet. Part A 2021, 185, 2496–2501. [Google Scholar] [CrossRef]
- Sen, G.; Jackson, T. Laminopathies: Should Wenckebach be a cause for concern? A case report. Eur. Heart J.—Case Rep. 2021, 5, ytab331. [Google Scholar] [CrossRef]
- Leone, M.P.; Palumbo, P.; Saenen, J.; Mastroianno, S.; Castellana, S.; Amico, C.; Mazza, T.; Potenza, D.R.; Petracca, A.; Castori, M.; et al. Phenotypic Variability of a Pathogenic PKP2 Mutation in an Italian Family Affected by Arrhythmogenic Cardiomyopathy and Juvenile Sudden Death: Considerations From Molecular Autopsy to Sport Restriction. Front. Cardiovasc. Med. 2021, 8, 635141. [Google Scholar] [CrossRef] [PubMed]
- Siskind, T.; Williams, N.; Sebastin, M.; Marion, R.; McDonald, T.V.; Walsh, C.; Sampson, B.; Tang, Y.; Clark, B.C. Genetic screening of relatives of decedents experiencing sudden unexpected death: Medical examiner’s office referrals to a multi-disciplinary cardiogenetics program. J. Community Genet. 2022, 13, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, J.; Kissel, C.K.; Bolliger, S.A.; Barbon, D.; Thali, M.J.; Kloiber, D.; Bode, P.K.; Kovacs, B.; Graf, U.; Maspoli, A.; et al. Benefits and outcomes of a new multidisciplinary approach for the management and financing of sudden unexplained death cases in a forensic setting in Switzerland. Forensic Sci. Int. 2022, 334, 111240. [Google Scholar] [CrossRef] [PubMed]
- Kraoua, L.; Jaouadi, H.; Allouche, M.; Achour, A.; Kaouther, H.; Ahmed, H.B.; Chaker, L.; Maazoul, F.; Ouarda, F.; Zaffran, S.; et al. Molecular autopsy and clinical family screening in a case of sudden cardiac death reveals ACTN2 mutation related to hypertrophic/dilated cardiomyopathy and a novel LZTR1 variant associated with Noonan syndrome. Mol. Genet. Genom. Med. 2022, 10, e1954. [Google Scholar] [CrossRef] [PubMed]
- Votýpka, P.; Krebsová, A.; Norambuena-Poustková, P.; Peldová, P.; Pohlová Kučerová, Š.; Kulvajtová, M.; Dohnalová, P.; Bílek, M.; Stufka, V.; Rücklová, K.; et al. Post-mortem genetic testing in sudden cardiac death and genetic screening of relatives at risk: Lessons learned from a Czech pilot multidisciplinary study. Int. J. Leg. Med. 2023, 137, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- «MedlinePlus: Genes». Consultato: 02 maggio 2024. [Online]. Available online: https://medlineplus.gov/genetics/gene/ (accessed on 28 March 2024).
- «GeneCards—Human Genes|Gene Database|Gene Search». Consultato: 02 maggio 2024. [Online]. Available online: https://www.genecards.org/ (accessed on 28 March 2024).
- Chugh, S.S.; Senashova, O.; Watts, A.; Tran, P.T.; Zhou, Z.; Gong, Q.; Titus, J.L.; Hayflick, S.J. Postmortem molecular screening in unexplained sudden death. J. Am. Coll. Cardiol. 2004, 43, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Stattin, E.-L.; Westin, I.M.; Cederquist, K.; Jonasson, J.; Jonsson, B.-A.; Mörner, S.; Norberg, A.; Krantz, P.; Wisten, A. Genetic screening in sudden cardiac death in the young can save future lives. Int. J. Leg. Med. 2016, 130, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, A.; Keyser, C.; Hollard, C.; Raul, J.S.; Muller, J.; Ludes, B. Targeted next generation sequencing application in cardiac channelopathies: Analysis of a cohort of autopsy-negative sudden unexplained deaths. Forensic Sci. Int. 2015, 254, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Lahrouchi, N.; Raju, H.; Lodder, E.M.; Papatheodorou, E.; Ware, J.S.; Papadakis, M.; Tadros, R.; Cole, D.; Skinner, J.R.; Crawford, J.; et al. Utility of Post-Mortem Genetic Testing in Cases of Sudden Arrhythmic Death Syndrome. J. Am. Coll. Cardiol. 2017, 69, 2134–2145. [Google Scholar] [CrossRef]
- Neubauer, J.; Lecca, M.R.; Russo, G.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W.; Haas, C. Exome analysis in 34 sudden unexplained death (SUD) victims mainly identified variants in channelopathy-associated genes. Int. J. Leg. Med. 2018, 132, 1057–1065. [Google Scholar] [CrossRef]
- Neubauer, J.; Wang, Z.; Rougier, J.-S.; Abriel, H.; Rieubland, C.; Bartholdi, D.; Haas, C.; Medeiros-Domingo, A. Functional characterization of a novel SCN5A variant associated with long QT syndrome and sudden cardiac death. Int. J. Leg. Med. 2019, 133, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Mahlke, N.; Dittmann, S.; Schulze-Bahr, E.; Ritz-Timme, S.; Hartung, B. Sudden unexpected cardiac death and postmortem identification of a novel RYR2 gene mutation. Int. J. Leg. Med. 2019, 133, 1835–1838. [Google Scholar] [CrossRef] [PubMed]
- Simons, E.; Labro, A.; Saenen, J.; Nijak, A.; Sieliwonczyk, E.; Vandendriessche, B.; Dąbrowska, M.; Van Craenenbroeck, E.M.; Schepers, D.; Van Laer, L.; et al. Molecular autopsy and subsequent functional analysis reveal de novo DSG2 mutation as cause of sudden death. Eur. J. Med. Genet. 2021, 64, 104322. [Google Scholar] [CrossRef] [PubMed]
- Scheiper-Welling, S.; Tabunscik, M.; Gross, T.E.; Jenewein, T.; Beckmann, B.M.; Niess, C.; Gradhand, E.; Wunder, C.; Schneider, P.M.; Rothschild, M.A.; et al. Variant interpretation in molecular autopsy: A useful dilemma. Int. J. Leg. Med. 2022, 136, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Avishay, D.M.; Jones, C.R.; Shaikh, J.D.; Kaur, R.; Aljadah, M.; Kichloo, A.; Shiwalkar, N.; Keshavamurthy, S. Sudden cardiac death: Epidemiology, pathogenesis and management. Rev. Cardiovasc. Med. 2021, 22, 147. [Google Scholar] [CrossRef] [PubMed]
- Del Duca, F.; Manetti, A.C.; Maiese, A.; Napoletano, G.; Ghamlouch, A.; Pascale, N.; Giorgio, B.; Paola, F.; Russa, R.L. Death Due to Anaphylactic Reaction: The Role of the Forensic Pathologist in an Accurate Postmortem Diagnosis. Medicina 2023, 59, 2184. [Google Scholar] [CrossRef] [PubMed]
- Oktay, V. The Definition of Sarcomeric and Non-Sarcomeric Gene Mutations in Hypertrophic Cardiomyopathy Patients: A Multicenter Diagnostic Study Across Türkiye. Anatol. J. Cardiol. 2023, 27, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Zekavati, A.; Syrris, P.; Hubank, M.; Giambartolomei, C.; Dalageorgou, C.; Jenkins, S.; McKenna, W.; Uk10k Consortium; Plagnol, V.; et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J. Med. Genet. 2013, 50, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef]
- Shirani, J.; Pick, R.; Roberts, W.C.; Maron, B.J. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J. Am. Coll. Cardiol. 2000, 35, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Lakdawala, N.K.; Tschöpe, C.; Klingel, K. Dilated cardiomyopathy: Causes, mechanisms, and current and future treatment approaches. Lancet 2023, 402, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Dec, G.W.; Fuster, V. Idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1994, 331, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.P.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Krahn, A.D.; Laksman, Z.; Sy, R.W.; Postema, P.G.; Ackerman, M.J.; Wilde, A.A.M.; Han, H.-C. Congenital Long QT Syndrome. JACC Clin. Electrophysiol. 2022, 8, 687–706. [Google Scholar] [CrossRef]
- Krahn, A.D.; Behr, E.R.; Hamilton, R.; Probst, V.; Laksman, Z.; Han, H.-C. Brugada Syndrome. JACC Clin. Electrophysiol. 2022, 8, 386–405. [Google Scholar] [CrossRef]
- Bjerregaard, P. Diagnosis and management of short QT syndrome. Heart Rhythm 2018, 15, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Orphanet: Tachicardia Ventricolare Polimorfa Catecolaminergica. Available online: https://www.orpha.net/it/disease/detail/3286?name=cpvt&mode=name (accessed on 2 April 2024).
- Fellmann, F.; Van El, C.G.; Charron, P.; Michaud, K.; Howard, H.C.; Boers, S.N.; Clarke, A.J.; Duguet, A.-M.; Forzano, F.; Kauferstein, S.; et al. European recommendations integrating genetic testing into multidisciplinary management of sudden cardiac death. Eur. J. Hum. Genet. 2019, 27, 1763–1773. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | n. | Age (Average) | Sex | Gene | Disease |
|---|---|---|---|---|---|
| Van Driest et al. (2002) [8] | 1 | 8 | F | TPM1 | HCM |
| Di Gioia et al. (2006) [9] | 100 | 2–40 (30.3) | M (69) F (31) | RYR2, KVLQT1, HERG, SCN5A | HCM, DCM, ARVC, Channelopathy |
| Larsen et al. (2011) [10] | 41 | 0–40 | M (/) F (/) | MYH7, MYBPC3, TNNI3, TNNT2, MYL2, MYL3, LMNA, PKP2, DSP, DSG2, DCS2, JUP, TMEM43 | HCM, DCM, ARVC |
| Campuzano et al. (2014) [11] | 29 | 21 days—14 (3.29) | M (14) F (15) | SCN5A, KCNQ1, KCNH2, KCNE1, KCNE2, KCNE3, RyR2, MYBPC3, MYH7, PKP2, DSC2, DSP, LMNA | ACM, HCM, DCM, LQTS, SQTS, BrS |
| Santori et al. (2015) [12] | 41 | 18 weeks (SIDS:38) and 3 years (SADS:3) | M/F (74%, SIDS), M/F (67%, SADS) | MYBPC3, MYH6, JUP, LDB3, DSC2, TTN, MYLK2, AKAP9, FBN1, SCN5A, MYH7, RYR2, DSG2, DES, GLA, KCNE1L, MYL2, DSP, RANGRF, DMD, TNNT2, BAG3, SCN1B, RBM20 | HCM, DCM, ACM, FABRY DISEASE, CHANNELOPATHY |
| Narula et al. (2015) [13] | 14 | 1.3–29 (17.4) | M (8) F (6) | KCNQ1, KCNH2, SCN5A, RYR2, MYH7, MYBC3, TTN, CACNA1C, JPH2, VLC | HCM, DCM, ACM, Channelopathy |
| Hertz et al. (2015) [14] | 72 | 1–50 (41) | M (50) F (22) | SCN5A, TRPM4, LDB3, LMNA, HCN4, DSP, MYBPC3, TTN, RYR2, NPPA, CACNA1C, MYH7, KCNA5, KCNQ1, KCNH2, CASQ2, MYH6, NEXN, AKAP9 | HCM, ARCV |
| Neubauer et al. (2016) [15] | 5 | 19–38 (29) | M (1) F (4) | DCHS1, TGFB2, GJD4, JPH2, DSP, KCNH2, MYH7, RANGRF, KCNQ1, SCN10A, SCN5A | MARFAN (MITRAL VALVE PROLAPSE), LQTS, ARVC, METABOLIC IMBALANCE |
| Hellenthal et al. (2017) [16] | 10 | 19–40 | M (5) F (5) | TTN, BAG3, DSG2, KCNH2, MYPN, CACNA1C, PRDM16, KCNE3, ABCC9, CACNB2, SCN5A, AKAP9, LAMA4, DSP, TNNT2 | HCM, ARVC, DCM |
| Campuzano et al. (2017) [17] | 52 | 14–50 (37.19) | M (48) F (4) | RYR2, TTN, ANK2, TNNT2, MYH7, PKP2, MYBPC3, DSC2, CACNA1C, NEXN, CACNB2, TNNI3, DSG2, CSRP3, HCN4, LAMP2 SGCD, CAV3, KCNH2, DSP, ANK2, MYH6, BAG3, HCN4, RBM20 | ACM |
| Andersen et al. (2019) [18] | 13 | 0 (SUDS) 29–50 (44, SADS) | M (10) F (3) | MYL2, MYL7, MYL4, PLN, FABP3, MYL3, TNNT2, ACTC1, TNNI3, MYH7, TTN, TNNC1, MB, DES, CKM, ACTA1, ANKRD1 | DCM, HCM |
| Marey et al. (2020) [19] | 35 | 21 days—72 (25.5) | M (18) F (17) | DSP, LMNA, TNNT2, TNNI3, MYBPC3, TTR | ARVC, HCM, DCM, RCM, LVNC |
| Grassi et al. (2020) [20] | 1 | 7 | F (1) | MYBPC3, MYH7, TNNI3, TNNT2, BAG3, LMNA, TTN, DSC2, DSP, PKP2, DSG2 | HCM, DCM, ACM |
| Fernlund et al. (2020) [21] | 10 | 0.1–24 (12.5) | M (8) F (2) | MYH7, ABCC9, FLNC, MYBPC3, PGM1, RBM20, ALPK3 | HCM |
| Fahed et al. (2020) [22] | 57 | 7–64 | M (29) F (28) | TNNI3 | HCM |
| Webster et al. (2021) [23] | 2 | 7 months and 1 month | M (2) | C1QPB | HCM |
| Sen et al. (2021) [24] | 1 | 42 | M (1) | LMNA | DCM |
| Leone et al. (2021) [25] | 1 | 13 | M (1) | PKP2 | ACM |
| Siskind et al. (2022) [26] | 15 | 2 days—57 (15) | M (/) F (/) | RYR2, PRDM16, SCN10A, MYH7, MYBPC3, KCNH2, CASQ2, TRDN, SCN5A, MYH6, KCNA5, CACNA1C, MYLK2 | CPVT, LQTS, ARVC, DCM, LVNC, BrS, HCM |
| Neubauer et al. (2022) [27] | 10 | 6–55 (33.1) | M (9) F (5) | AKAP9, FHOD3, RBM20, LMNA, DSP, APOB, ABCC9, CDH2, JUP, MYBC3, RYR2, SCN10A, ZIC3, UBR4, ABCC6, GAA, MYOM1, FLNC, LDB3, LRRC10, MYH7, MYH11, SLC4A3, ACADM, LDB3, SLC22A5, AGPAT2, DSC2, SCN10A, TTN | ACM, DCM, SQTS, LQTS |
| Kraoua et al. (2022) [28] | 2 | 28—3 and 10 months | M (2) | ACTN2 | HCM |
| Votýpka et al. (2023) [29] | 100 | (33.3) | M (71) F (29) | GLA, KCNQ1, MYBPC3, SCN5A, RBM20, FHL1, TTN, FLNC, MYPN, COL3A1, TGFBR1, KCNH2, RYR2, TNNT2, DES, DSP, CTNNA3, PRKAG2, DPP6, LMNA, KCNE1 | HCM, DCM, ARVC, LQTS |
| Genes | Functions | Sarcomeric or Not |
|---|---|---|
| LMNA | Provides instructions for making several slightly different proteins called lamins. The two major proteins produced from this gene, lamin A and lamin C, are made in most of the body’s cells. Lamins A and C are supporting (scaffolding) components of the nuclear envelope, which is a structure that surrounds the nucleus in cells. | Not sarcomeric |
| C1QPB | Is believed to be a multifunctional and multicompartmental protein involved in inflammation and infection processes, ribosome biogenesis, protein synthesis in mitochondria, regulation of apoptosis, transcriptional regulation, and pre-mRNA splicing. | Not sarcomeric |
| RYR2 | Provides instructions for making a protein called ryanodine receptor 2. This protein is part of a family of ryanodine receptors, which form channels that transport positively charged calcium atoms (calcium ions) within cells. | Not sarcomeric |
| PKP2 | Provides instructions for making a protein called plakophilin 2. This protein is found primarily in cells of the myocardium, which is the muscular wall of the heart. | Not sarcomeric |
| SCN5A | Belongs to a family of genes that provide instructions for making sodium channels. These channels open and close at specific times to control the flow of positively charged sodium atoms (sodium ions) into cells. | Not sarcomeric |
| KCNQ1 | Belongs to a large family of genes that provide instructions for making potassium channels. These channels, which transport positively charged atoms (ions) of potassium out of cells, play key roles in a cell’s ability to generate and transmit electrical signals. | Not sarcomeric |
| KCNH2 | Belongs to a large family of genes that provide instructions for making potassium channels. These channels, which transport positively charged atoms (ions) of potassium out of cells, play key roles in a cell’s ability to generate and transmit electrical signals. | Not sarcomeric |
| KCNE1 | Belongs to a large family of genes that provide instructions for making potassium channels. These channels, which transport positively charged atoms (ions) of potassium out of cells, play key roles in a cell’s ability to generate and transmit electrical signals. | Not sarcomeric |
| MYBPC3 | Delivers instructions for making cardiac myosin binding protein C (cardiac MyBP-C). | Sarcomeric |
| MYH7 | Provides instructions for making a protein known as the beta (b)myosin heavy chain (sarcomere). | Sarcomeric |
| TNNT2 | Provides instructions for making a protein called cardiac troponin T, which is found solely in the heart (cardiac) muscle. | Sarcomeric |
| TNNI3 | Provides instructions for making a protein called cardiac troponin I, which is found solely in the heart (cardiac) muscle. | Sarcomeric |
| TTN | Provides instructions for making a very large protein called titin. This protein plays an important role in skeletal muscles, which the body uses for movement, and in heart (cardiac) muscle. | Sarcomeric |
| ACTN2 | Encodes alpha-actinin-2, a protein expressed in human cardiac and skeletal muscle. The protein, located in the sarcomere Z-disk, functions as a link between the anti-parallel actin filaments. | Sarcomeric |
| TPM1 | Essential sarcomeric component, stabilising the thin filament and facilitating actin’s interaction with myosin. | Sarcomeric |
| DSP | Provides instructions for making a protein called desmoplakin. This protein is found primarily in cells of the heart and skin, where it is a major component of specialised structures called desmosomes. | Sarcomeric |
| DSG2 | Encodes a member of the desmoglein family and cadherin cell adhesion molecule superfamily of proteins. Desmogleins are calcium-binding transmembrane glycoprotein components of desmosomes, cell–cell junctions between epithelial, myocardial, and other cell types. | Sarcomeric |
| DSC2 | Provides instructions for making a protein called desmocollin-2. This protein is found in many tissues, although it appears to be particularly important in the heart muscle and skin. Desmocollin-2 is a major component of specialized structures called desmosomes. | Sarcomeric |
| References | n. | Age (Average) | Sex | Gene | Disease |
|---|---|---|---|---|---|
| Chugh et al. (2004) [32] | 12 | - | - | KCNQ1 (KVLQT1), KCNH2 (HERG), SCN5A, KCNE1, and KCNE2 | LQTS, WPW, BrS, CPVT |
| Di Gioia et al. (2006) [8] | 100 | 2–40 (30.3) | M (69) F (31) | RYR2, KVLQT1, HERG, SCN5A | HCM, DCM, ARVC, CAD, Channelopathy |
| Campuzano et al. (2014) [10] | 29 | 21 days—14 (3.29) | M (14) F (15) | SCN5A, KCNQ1, KCNH2, KCNE1, KCNE2, KCNE3, RyR2 | ACM, HCM, DCM, LQTS, SQTS, BrS |
| Santori et al. (2015) [11] | 41 | 18 weeks (SIDS:38) and 3 years (SADS:3) | M/F (74%, SIDS), M/F (67%, SADS) | MYBPC3, MYH6, JUP, LDB3, DSC2, TTN, MYLK2, AKAP9, FBN1, SCN5A, MYH7, RYR2, DSG2, DES, GLA, KCNE1L, MYL2, DSP, RANGRF, DMD, TNNT2, BAG3, SCN1B, RBM20 | HCM, DCM, ACM, FABRY DISEASE, channelopathy |
| Stattin et al. (2015) [33] | 15 | (15) | M (10) F (5) | KCNQ1, KCNH2, RYR2, KCNE1, SCN5A | LQTS, CPVT, BrS, SQTS |
| Hertz et al. (2015) [13] | 72 | 1–50 (41) | M (50) F (22) | SCN5A, TRPM4, LDB3, LMNA, HCN4, DSP, MYBPC3, TTN, RYR2, NPPA, CACNA1C, MYH7, KCNA5, KCNQ1, KCNH2, CASQ2, MYH6, NEXN, AKAP9 | HCM, DCM, ARCV, LQTS, BrS |
| Narula et al. (2015) [12] | 14 | 1.3–29 (17.4) | M (8) F (6) | KCNQ1, KCNH2, SCN5A, RYR2, MYH7, MYBC3, TTN, CACNA1C, JPH2, VLC | HCM, DCM, ACM, Channelopathy |
| Farrugia et al. (2015) [34] | 16 | 2m—34 | M (8) F (8) | KCNH2, SCN5A, ANK2, RYR2, KCNE1, CASQ2 | LQTS, BrS, CPVT |
| Neubauer et al. (2016) [14] | 5 | 19–38 (29) | M (1) F (4) | DCHS1, TGFB2, GJD4, JPH2, DSP, KCNH2, MYH7, RANGRF, KCNQ1, SCN10A, SCN5A | MARFAN (MITRAL VALVE PROLAPSE, LQTS, ARVC, METABOLIC IMBALANCE |
| Hellenthal et al. (2017) [15] | 10 | 19–40 | M (5) F (5) | TTN, BAG3, DSG2, KCNH2, MYPN, CACNA1C, PRDM16, KCNE3, ABCC9, CACNB2, SCN5A, AKAP9, LAMA4, DSP, TNNT2 | HCM, ARVC, DCM, LQTS |
| Campuzano et al. (2017) [16] | 52 | 14–50 (37.19) | M (48) F (4) | RYR2, TTN, ANK2, TNNT2, MYH7, PKP2, MYBPC3, DSC2, CACNA1C, NEXN, CACNB2, TNNI3, DSG2, CSRP3, HCN4, LAMP2, SGCD, CAV3, KCNH2, DSP, ANK2, MYH6, BAG3, PKP2, HCN4, RBM20 | ACM |
| Lahrouchi et al. (2017) [35] | 302 | (24) | M (196) F (106) | KCNQ1, KCNH2, SCN5A, RYR2 | CPVT, LQTS |
| Neubauer et al. (2018) [36] | 34 | 1–63 | M (26) F (8) | RYR2, ACAD9, AKAP9, SEMA3A | CPVT, BrS, LQTS |
| Neubauer et al. (2019) [37] | 1 | 19 | F (1) | SCN5A | LQTS |
| Mahlke et al. (2019) [38] | 1 | 13 | F (1) | RYR2 | CPVT |
| Simons et al. (2021) [39] | 1 | 49 | M (1) | KCNQ1, DSG2 | LQTS |
| Neubauer et al. (2022) [26] | 10 | 6–55 (33.1) | M (9) F (5) | AKAP9, FHOD3, RBM20, LMNA, DSP, APOB, ABCC9, CDH2, JUP, MYBC3, SCN10A, ZIC3, UBR4, ABCC6, GAA, MYOM1, FLNC, LDB3, LRRC10, MYH7, MYH11, RYR2, SLC4A3, ACADM, LDB3, SLC22A5, AGPAT2, DSC2, SCN10A, TTN | ACM, DCM, SQTS, LQTS |
| Siskind et al. (2022) [25] | 15 | 2 days—57 (15) | M (/) F (/) | RYR2, PRDM16, SCN10A, MYH7, MYBPC3, KCNH2, CASQ2, TRDN, SCN5A, MYH6, KCNA5, CACNA1C, MYLK2 | CPVT, LQTS, ARVC, DCM, LVNC, BrS, HCM |
| Scheiper-Welling et al. (2022) [40] | 56 | 1–50 | M (/) F (/) | KCNH2, SCN5A, KCNQ1, MYBC3, VCL, JUP | ARRHYTMIC DEATH |
| Votýpka et al. (2023) [28] | 100 | (33.3) | M (71) F (29) | GLA, KCNQ1, MYBPC3, SCN5A, RBM20, FHL1, TTN, FLNC, MYPN, COL3A1, TGFBR1, KCNH2, RYR2, TNNT2, DES, DSP, CTNNA3, PRKAG2, DPP6, LMNA, KCNE1 | HCM, DCM, ARVC, LQTS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Duca, F.; Ghamlouch, A.; Manetti, A.C.; Napoletano, G.; Sonnini, E.; Treves, B.; De Matteis, A.; La Russa, R.; Sheppard, M.N.; Fineschi, V.; et al. Sudden Cardiac Death, Post-Mortem Investigation: A Proposing Panel of First Line and Second Line Genetic Tests. J. Pers. Med. 2024, 14, 544. https://doi.org/10.3390/jpm14050544
Del Duca F, Ghamlouch A, Manetti AC, Napoletano G, Sonnini E, Treves B, De Matteis A, La Russa R, Sheppard MN, Fineschi V, et al. Sudden Cardiac Death, Post-Mortem Investigation: A Proposing Panel of First Line and Second Line Genetic Tests. Journal of Personalized Medicine. 2024; 14(5):544. https://doi.org/10.3390/jpm14050544
Chicago/Turabian StyleDel Duca, Fabio, Alessandro Ghamlouch, Alice Chiara Manetti, Gabriele Napoletano, Elena Sonnini, Biancamaria Treves, Alessandra De Matteis, Raffaele La Russa, Mary N. Sheppard, Vittorio Fineschi, and et al. 2024. "Sudden Cardiac Death, Post-Mortem Investigation: A Proposing Panel of First Line and Second Line Genetic Tests" Journal of Personalized Medicine 14, no. 5: 544. https://doi.org/10.3390/jpm14050544
APA StyleDel Duca, F., Ghamlouch, A., Manetti, A. C., Napoletano, G., Sonnini, E., Treves, B., De Matteis, A., La Russa, R., Sheppard, M. N., Fineschi, V., & Maiese, A. (2024). Sudden Cardiac Death, Post-Mortem Investigation: A Proposing Panel of First Line and Second Line Genetic Tests. Journal of Personalized Medicine, 14(5), 544. https://doi.org/10.3390/jpm14050544