
Elucidating the Impact of Deleterious Mutations on IGHG1 and Their Association with Huntington’s Disease





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
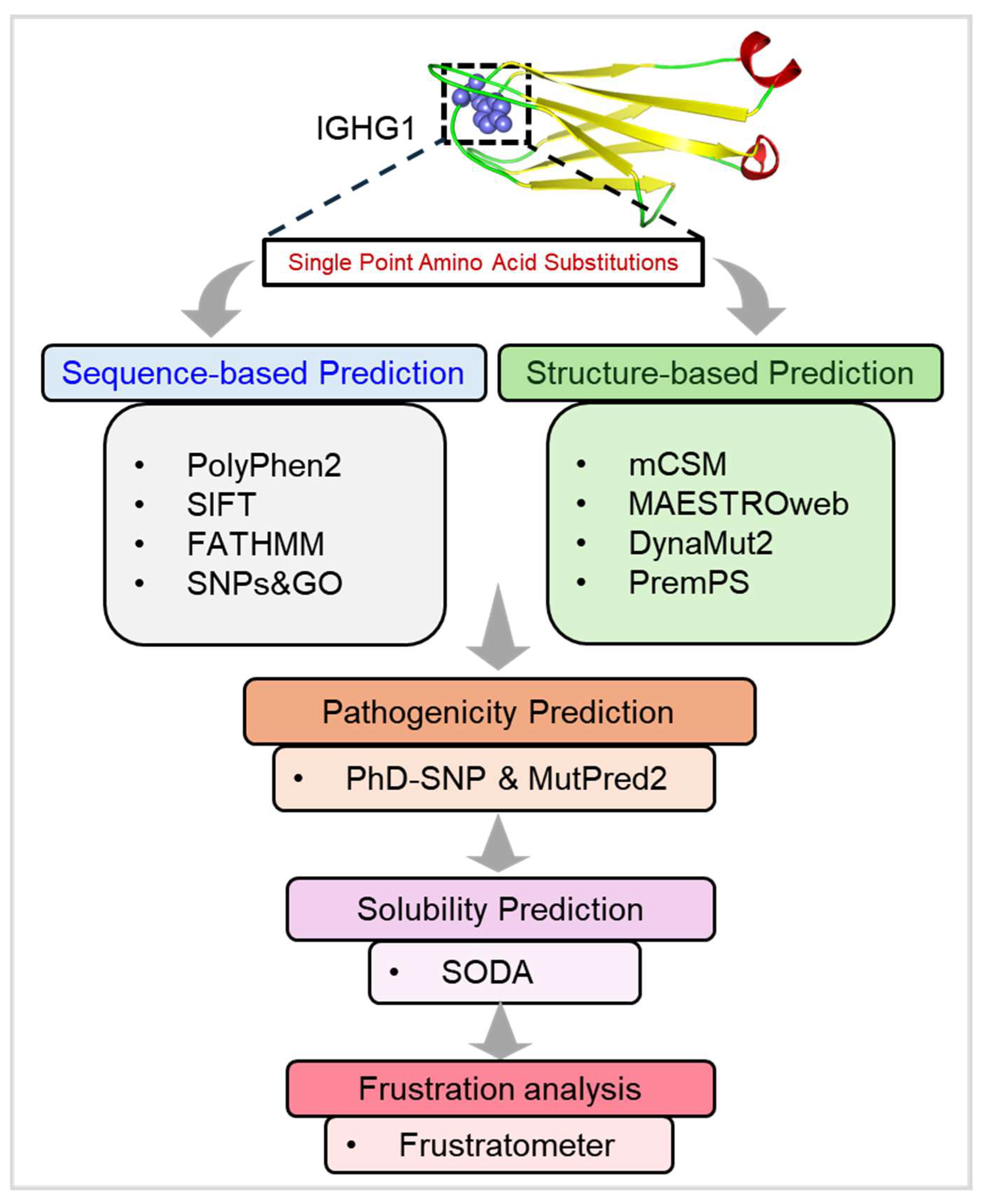
2. Materials and Methods
2.1. Data Retrieval and Web Resources
2.2. Sequence-Based Predictions
2.2.1. PolyPhen2
2.2.2. SIFT
2.2.3. FATHMM
2.2.4. SNPs&GO
2.3. Structure-Based Predictions
2.3.1. mCSM
2.3.2. MAESTROweb
2.3.3. PremPS
2.3.4. DynaMut2
2.4. Pathogenicity Prediction
2.4.1. PhD-SNP
2.4.2. MutPred2
2.5. Aggregation Propensity Analysis
2.6. Residual Frustration Analysis
3. Results
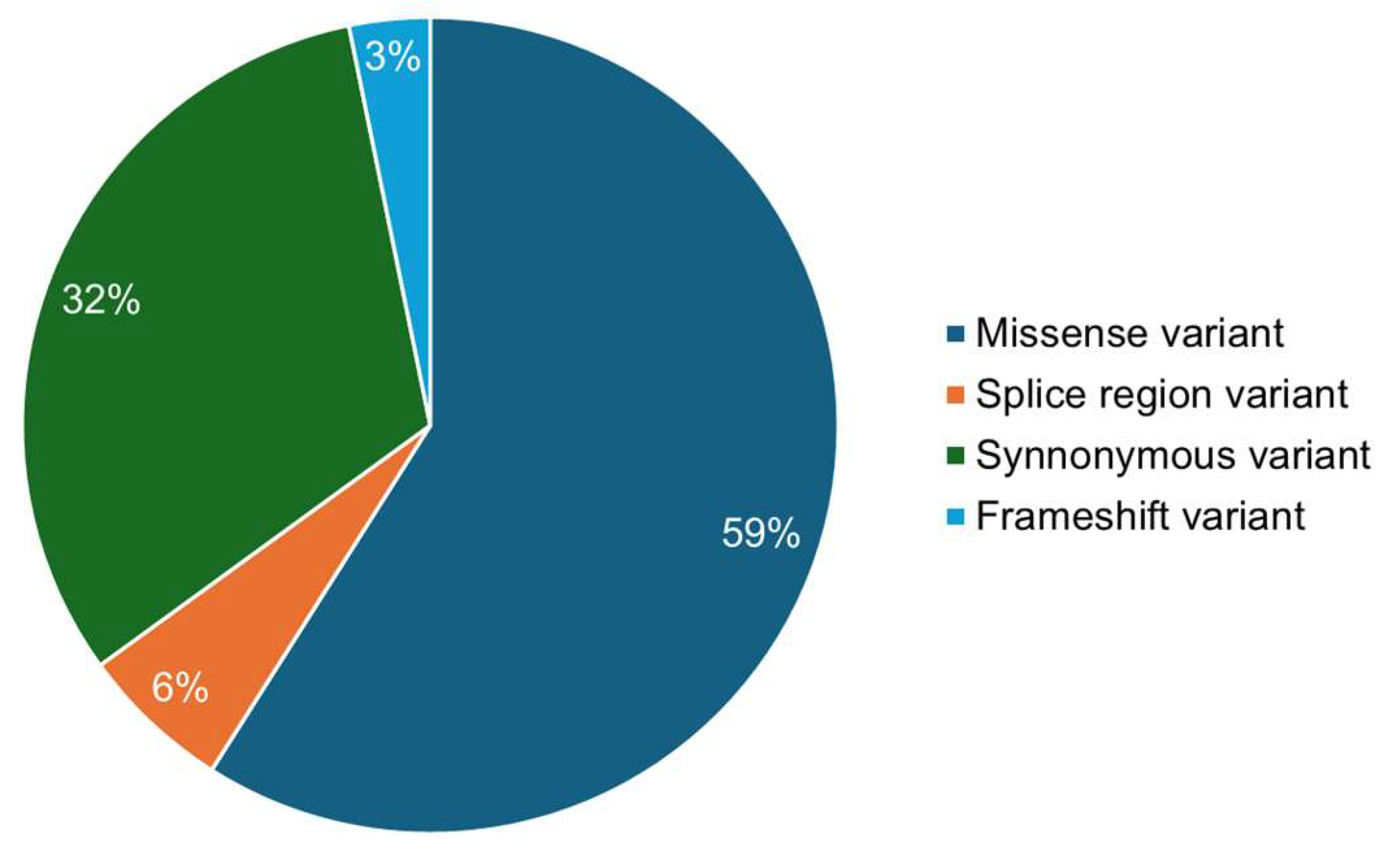
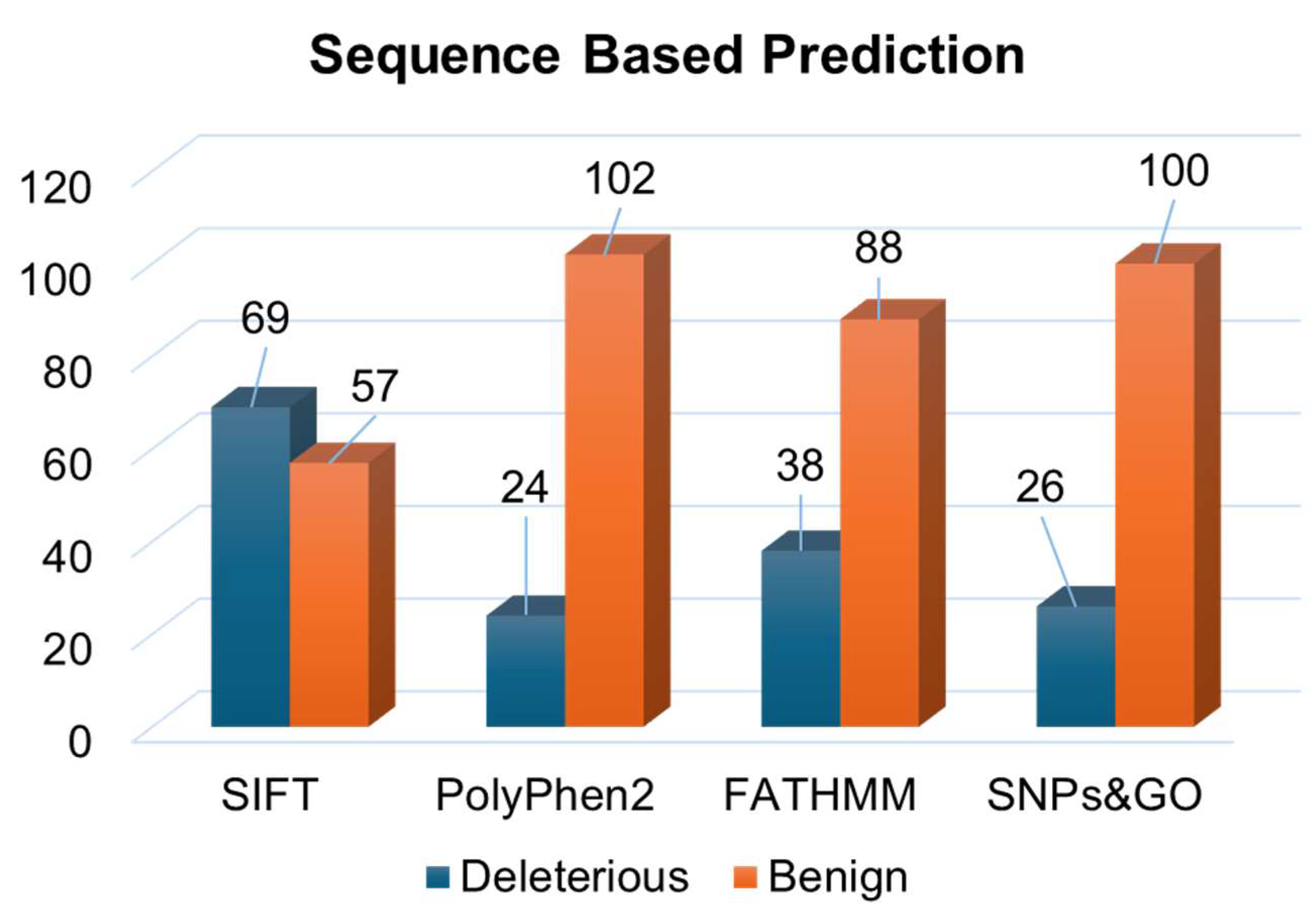
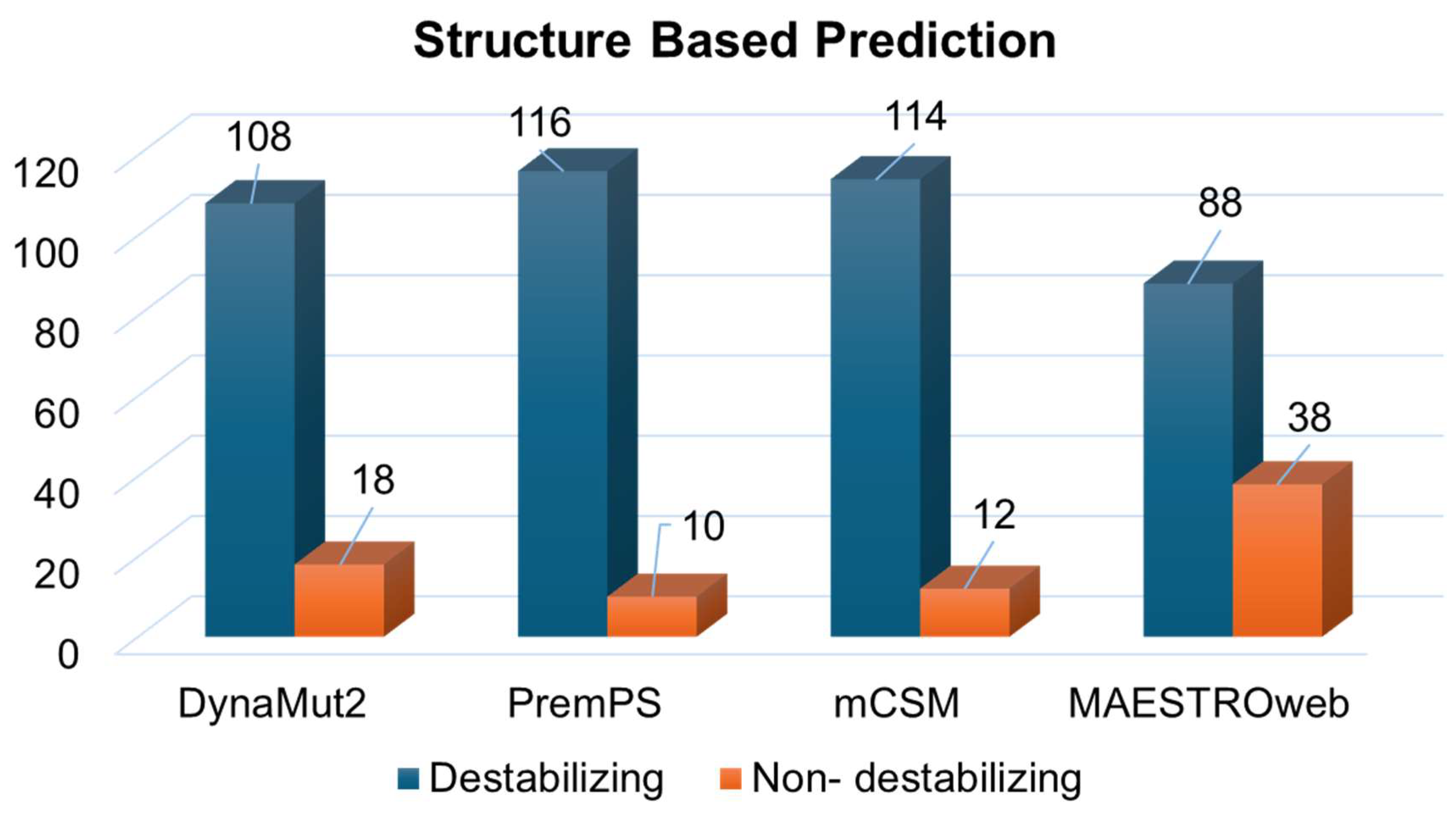
3.1. Deleterious Mutations from Sequence and Structure-Based Approaches
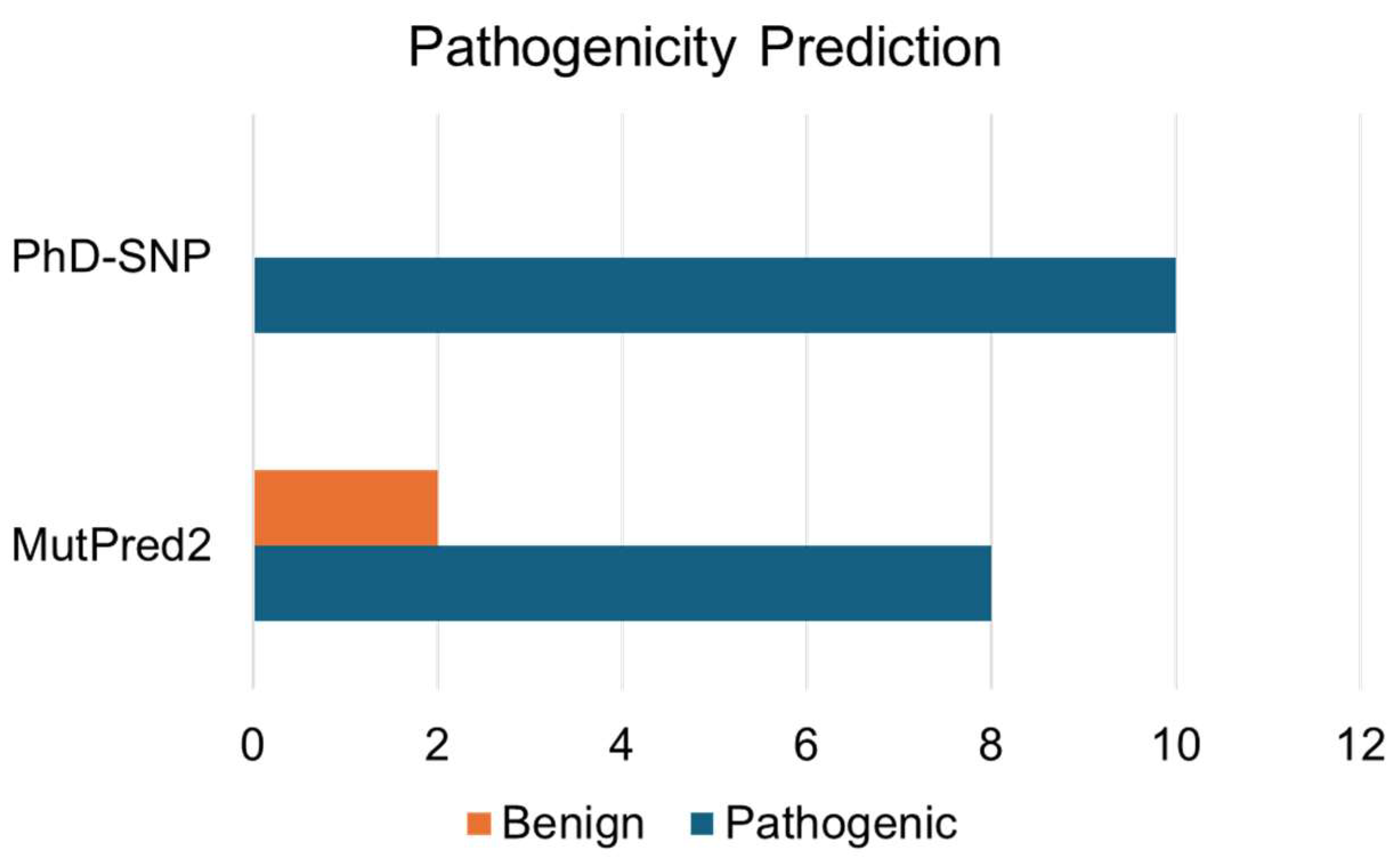
3.2. Identification of Disease-Associated Mutations
3.3. Residual Frustration Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Dayalu, P.; Albin, R.L. Huntington disease: Pathogenesis and treatment. Neurol. Clin. 2015, 33, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and incidence of Huntington’s disease: An updated systematic review and meta-analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, K.M.; Schwarz, A.J.; Turner, E.C.; Pustina, D.; Gantman, E.C.; Gordon, M.F.; Joules, R.; Mullin, A.P.; Scahill, R.I.; Georgiou-Karistianis, N. Volumetric MRI-based biomarkers in Huntington’s disease: An evidentiary review. Front. Neurol. 2021, 12, 712555. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pernaute, R.; Kunig, G.; del Barrio Alba, A.; De Yebenes, J.; Vontobel, P.; Leenders, K. Bradykinesia in early Huntington’s disease. Neurology 2000, 54, 119. [Google Scholar] [CrossRef] [PubMed]
- Dumas, E.M.; Van Den Bogaard, S.; Middelkoop, H.; Roos, R. A review of cognition in Huntington’s disease. Front. Biosci. (Sch. Ed.) 2013, 5, 1–18. [Google Scholar]
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral changes in Huntington disease. Cogn. Behav. Neurol. 2001, 14, 219–226. [Google Scholar]
- Zielonka, D.; Mielcarek, M.; Landwehrmeyer, G.B. Update on Huntington’s disease: Advances in care and emerging therapeutic options. Park. Relat. Disord. 2015, 21, 169–178. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.-P.G.; Faull, R.L. The neuropathology of Huntington’s disease. In Behavioral Neurobiology of Huntington’s Disease and Parkinson’s Disease; Springer: Berlin/Heidelberg, Germany, 2015; pp. 33–80. [Google Scholar]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- MacDonald, M.; Duyao, M.; Calzonetti, T.; Auerbach, A.; Ryan, A.; Barnes, G.; White, J.; Auerbach, W.; Vonsattel, J.-P.; Gusella, J. Targeted inactivation of the mouse Huntington’s disease gene homolog Hdh. Cold Spring Harb. Symp. Quant. Biol. 1996, 61, 627–638. [Google Scholar] [PubMed]
- Monckton, D.G. The contribution of somatic expansion of the CAG repeat to symptomatic development in Huntington’s disease: A historical perspective. J. Huntington’s Dis. 2021, 10, 7–33. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Lee, J.-M.; Gusella, J.F. Huntington’s disease genetics: Implications for pathogenesis. In Huntington’s Disease; Elsevier: Amsterdam, The Netherlands, 2024; pp. 57–84. [Google Scholar]
- Christodoulou, C.C.; Papanicolaou, E.Z. Omics and Network-Based Approaches in Understanding HD Pathogenesis; IntechOpen: London, UK, 2024. [Google Scholar]
- Testa, C.M.; Jankovic, J. Huntington disease: A quarter century of progress since the gene discovery. J. Neurol. Sci. 2019, 396, 52–68. [Google Scholar] [CrossRef]
- Lee, J.-M.; Wheeler, V.C.; Chao, M.J.; Vonsattel, J.P.G.; Pinto, R.M.; Lucente, D.; Abu-Elneel, K.; Ramos, E.M.; Mysore, J.S.; Gillis, T. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.M.; Wild, E.J. Cerebrospinal fluid biomarkers for Huntington’s disease. J. Huntington’s Dis. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; McColgan, P.; Robertson, N.; Tabrizi, S.J.; Zetterberg, H.; Wild, E.J. Cerebrospinal fluid inflammatory biomarkers reflect clinical severity in Huntington’s disease. PLoS ONE 2016, 11, e0163479. [Google Scholar] [CrossRef]
- Lederman, M.; Weiss, A.; Chowers, I. Association of neovascular age-related macular degeneration with specific gene expression patterns in peripheral white blood cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 53–58. [Google Scholar] [CrossRef]
- Caron, N.S.; Haqqani, A.S.; Sandhu, A.; Aly, A.E.; Findlay Black, H.; Bone, J.N.; McBride, J.L.; Abulrob, A.; Stanimirovic, D.; Leavitt, B.R. Cerebrospinal fluid biomarkers for assessing Huntington disease onset and severity. Brain Commun. 2022, 4, fcac309. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, M. Inflammation Biomarkers in Huntington’s Disease. In Biomarkers for Huntington’s Disease: Improving Clinical Outcomes; Springer: Berlin/Heidelberg, Germany, 2023; pp. 277–304. [Google Scholar]
- Malaiya, S.; Cortes-Gutierrez, M.; Herb, B.R.; Coffey, S.R.; Legg, S.R.; Cantle, J.P.; Colantuoni, C.; Carroll, J.B.; Ament, S.A. Single-nucleus RNA-seq reveals dysregulation of striatal cell identity due to huntington’s disease mutations. J. Neurosci. 2021, 41, 5534–5552. [Google Scholar] [CrossRef]
- Pan, L.; Feigin, A. Huntington’s disease: New frontiers in therapeutics. Curr. Neurol. Neurosci. Rep. 2021, 21, 10. [Google Scholar] [CrossRef]
- Amir, M.; Ahmad, S.; Ahamad, S.; Kumar, V.; Mohammad, T.; Dohare, R.; Alajmi, M.F.; Rehman, T.; Hussain, A.; Islam, A. Impact of Gln94Glu mutation on the structure and function of protection of telomere 1, a cause of cutaneous familial melanoma. J. Biomol. Struct. Dyn. 2019, 1514–1524. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Kumar, V.; Mohammad, T.; Dohare, R.; Rehman, M.T.; Alajmi, M.F.; Hussain, A.; Ahmad, F.; Hassan, M.I. Structural and functional impact of non-synonymous SNPs in the CST complex subunit TEN1: Structural genomics approach. Biosci. Rep. 2019, 39, BSR20190312. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Kumar, V.; Mohammad, T.; Dohare, R.; Hussain, A.; Rehman, M.T.; Alam, P.; Alajmi, M.F.; Islam, A.; Ahmad, F. Investigation of deleterious effects of nsSNPs in the POT1 gene: A structural genomics-based approach to understand the mechanism of cancer development. J. Cell. Biochem. 2019, 120, 10281–10294. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R. WS-SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genom. 2013, 14, S6. [Google Scholar]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A web server for structure-based protein stability prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, H.; Zhang, N.; Zhu, Z.; Wang, S.; Li, M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput. Biol. 2020, 16, e1008543. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, R.; Capriotti, E.; Casadio, R. PhD-SNP: A web server for the prediction of human genetic diseases associated to missense single nucleotide polymorphisms. In EMBNET08; ITA: Rome, Italy, 2008; p. 78. [Google Scholar]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Canese, K.; Weis, S. PubMed: The bibliographic database. In The NCBI Handbook; National Center for Biotechnology Information: Bethesda, MD, USA, 2013; Volume 2. [Google Scholar]
- Navarro, S.; Ventura, S. Computational methods to predict protein aggregation. Curr. Opin. Struct. Biol. 2022, 73, 102343. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Walsh, I.; Minervini, G.; Tosatto, S.C. FELLS: Fast estimator of latent local structure. Bioinformatics 2017, 33, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, D.U.; Komives, E.A.; Wolynes, P.G. Frustration in biomolecules. Q. Rev. Biophys. 2014, 47, 285–363. [Google Scholar] [CrossRef] [PubMed]
- Jenik, M.; Parra, R.G.; Radusky, L.G.; Turjanski, A.; Wolynes, P.G.; Ferreiro, D.U. Protein frustratometer: A tool to localize energetic frustration in protein molecules. Nucleic Acids Res. 2012, 40, W348–W351. [Google Scholar] [CrossRef]
- Amir, M.; Mohammad, T.; Kumar, V.; Alajmi, M.F.; Rehman, M.T.; Hussain, A.; Alam, P.; Dohare, R.; Islam, A.; Ahmad, F. Structural analysis and conformational dynamics of STN1 gene mutations involved in coat plus syndrome. Front. Mol. Biosci. 2019, 6, 41. [Google Scholar] [CrossRef]
- Paladin, L.; Piovesan, D.; Tosatto, S.C. SODA: Prediction of protein solubility from disorder and aggregation propensity. Nucleic Acids Res. 2017, 45, W236–W240. [Google Scholar] [CrossRef]
- Ciurea, A.V.; Mohan, A.G.; Covache-Busuioc, R.-A.; Costin, H.-P.; Glavan, L.-A.; Corlatescu, A.-D.; Saceleanu, V.M. Unraveling Molecular and Genetic Insights into Neurodegenerative Diseases: Advances in Understanding Alzheimer’s, Parkinson’s, and Huntington’s Diseases and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 10809. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.G. Late-onset neurodegenerative diseases—The role of protein insolubility. J. Anat. 2000, 196, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wanker, E.E. Protein aggregation and pathogenesis of Huntingtons disease: Mechanisms and correlations. Biol. Chem. 2000. [Google Scholar] [CrossRef]
- Anjum, F.; Joshia, N.; Mohammad, T.; Shafie, A.; Alhumaydhi, F.A.; Aljasir, M.A.; Shahwan, M.J.; Abdullaev, B.; Adnan, M.; Elasbali, A.M. Impact of single amino acid substitutions in parkinsonism-associated deglycase-PARK7 and their association with Parkinson’s disease. J. Pers. Med. 2022, 12, 220. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafie, A.; Ashour, A.A.; Anjum, F.; Shamsi, A.; Hassan, M.I. Elucidating the Impact of Deleterious Mutations on IGHG1 and Their Association with Huntington’s Disease. J. Pers. Med. 2024, 14, 380. https://doi.org/10.3390/jpm14040380
Shafie A, Ashour AA, Anjum F, Shamsi A, Hassan MI. Elucidating the Impact of Deleterious Mutations on IGHG1 and Their Association with Huntington’s Disease. Journal of Personalized Medicine. 2024; 14(4):380. https://doi.org/10.3390/jpm14040380
Chicago/Turabian StyleShafie, Alaa, Amal Adnan Ashour, Farah Anjum, Anas Shamsi, and Md. Imtaiyaz Hassan. 2024. "Elucidating the Impact of Deleterious Mutations on IGHG1 and Their Association with Huntington’s Disease" Journal of Personalized Medicine 14, no. 4: 380. https://doi.org/10.3390/jpm14040380
APA StyleShafie, A., Ashour, A. A., Anjum, F., Shamsi, A., & Hassan, M. I. (2024). Elucidating the Impact of Deleterious Mutations on IGHG1 and Their Association with Huntington’s Disease. Journal of Personalized Medicine, 14(4), 380. https://doi.org/10.3390/jpm14040380