
Monitoring Genomic Structural Rearrangements Resulting from Gene Editing

Abstract
1. Introduction
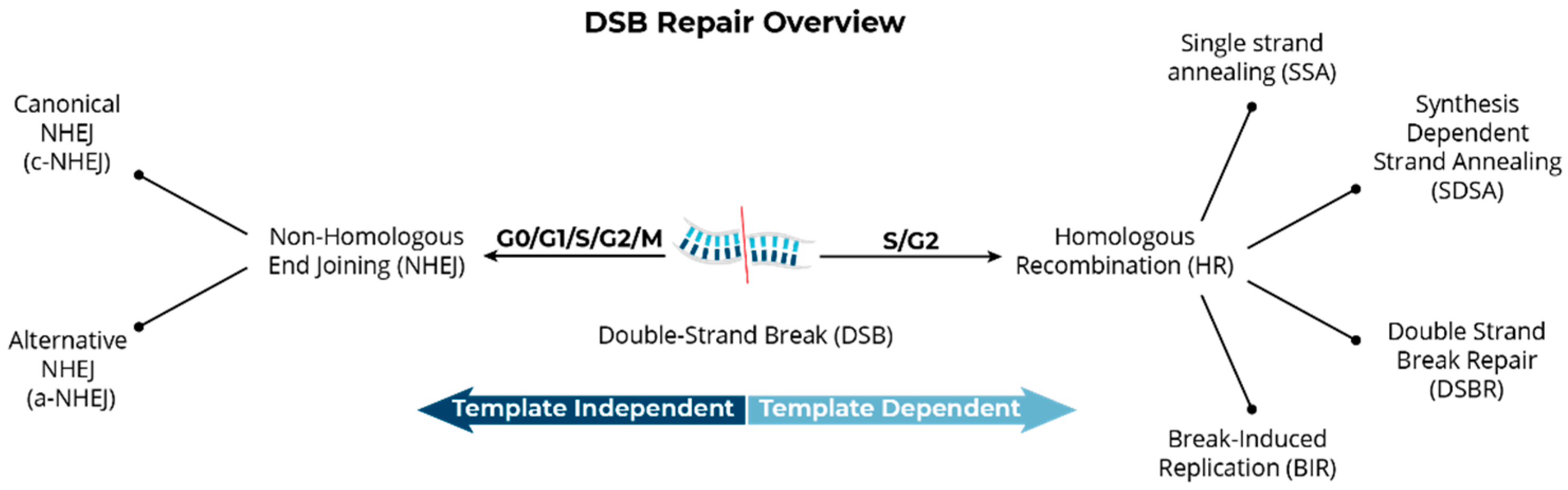
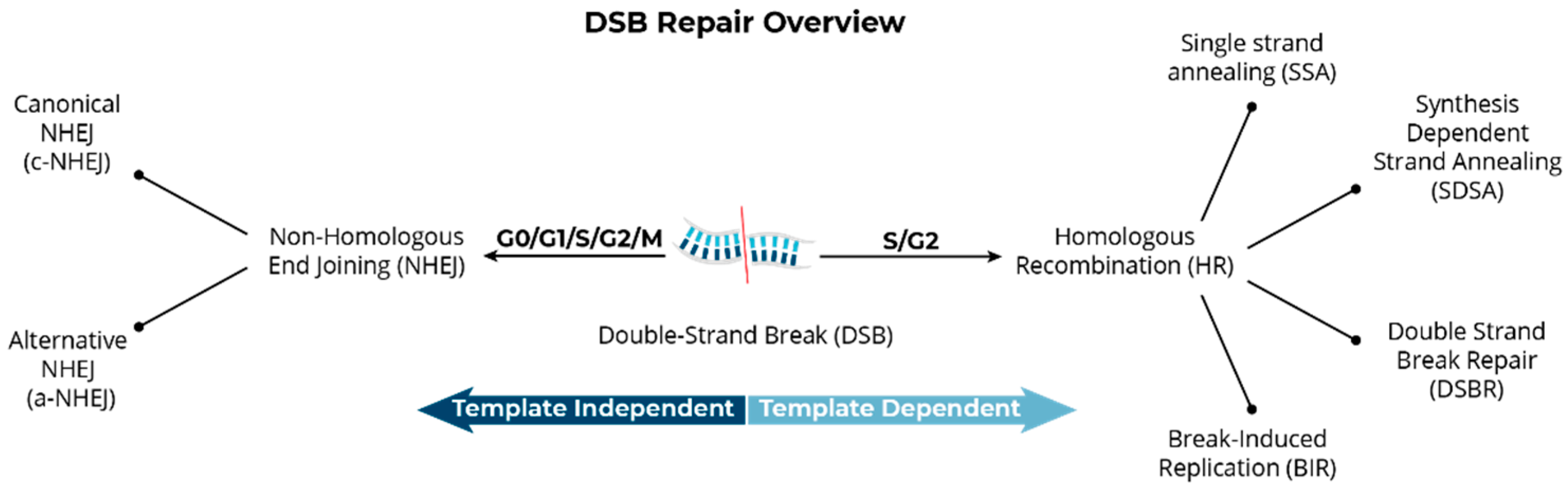
Structural Variants Arise from the Mis-Repair of DNA Double-Strand Breaks
2. Materials and Methods
3. Results
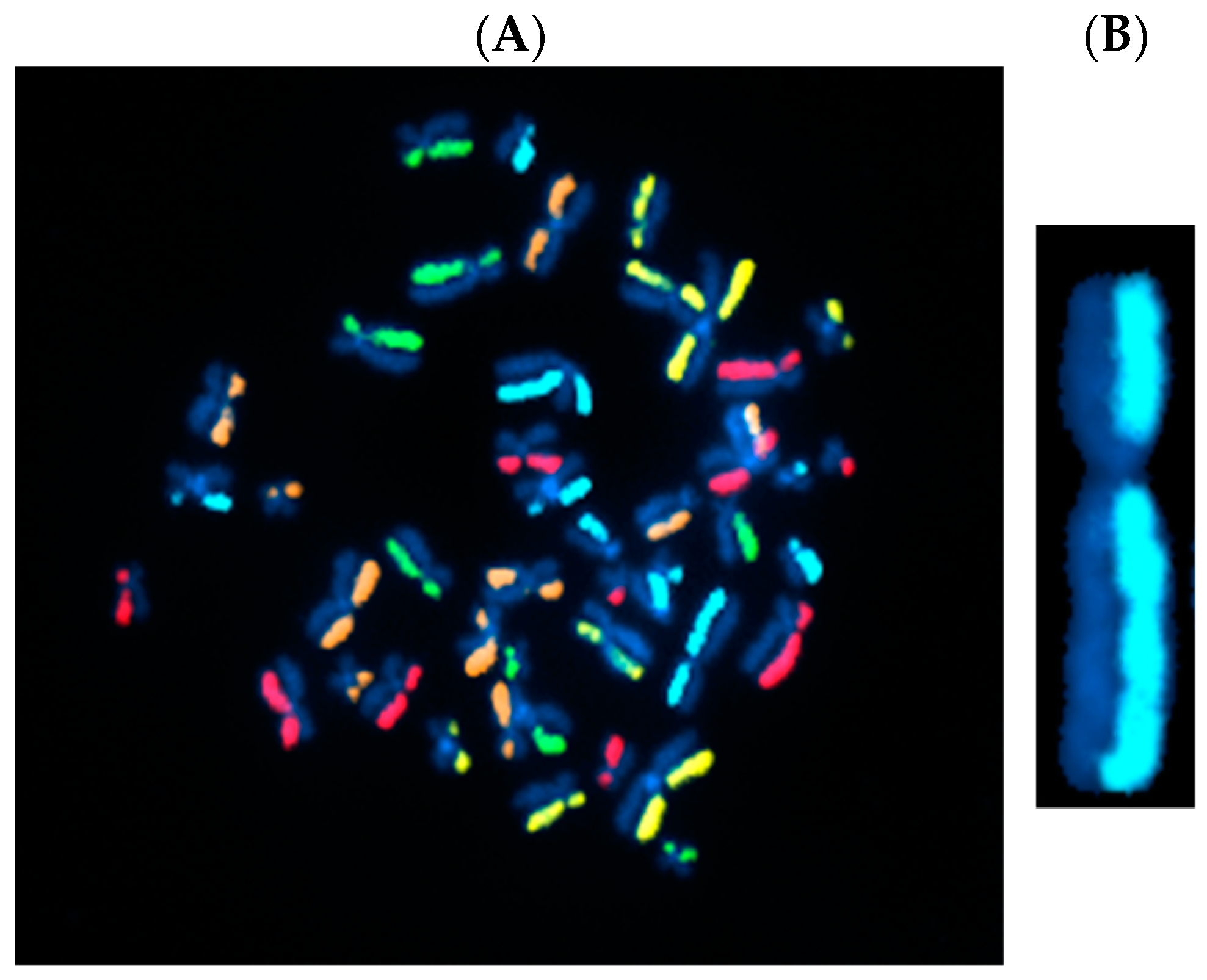
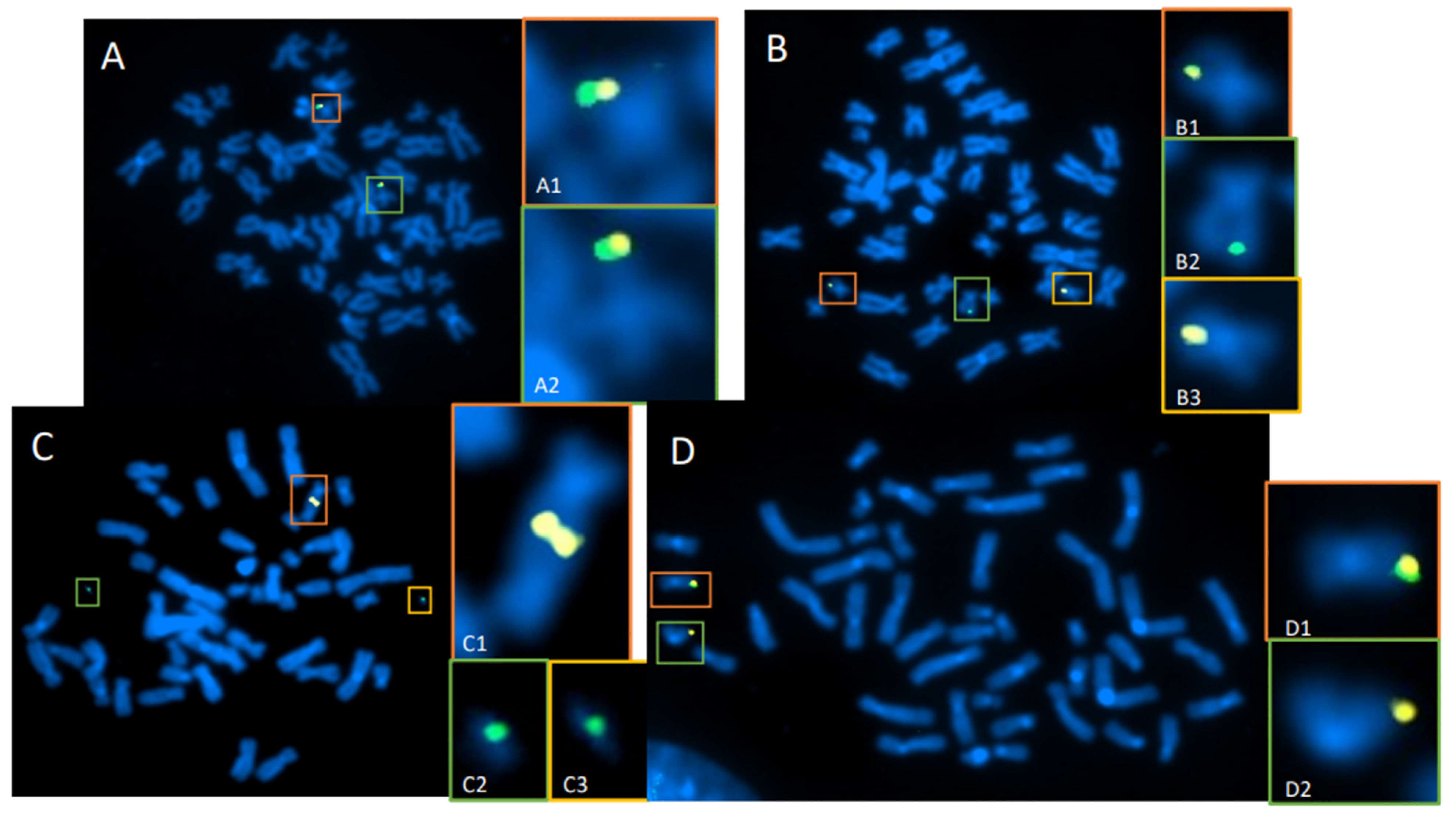
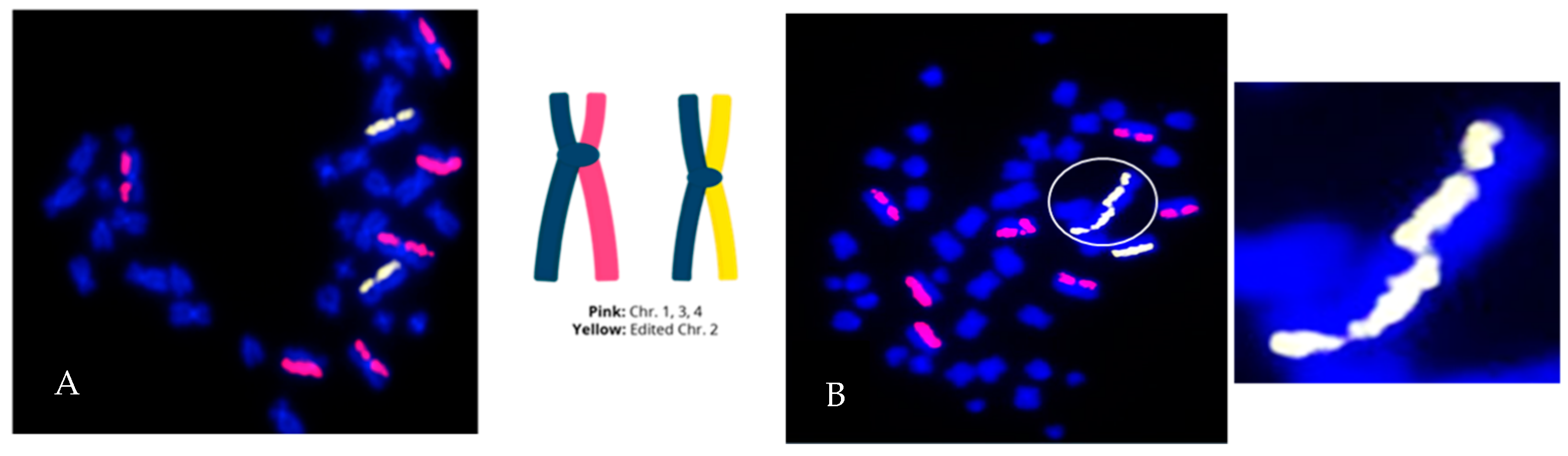
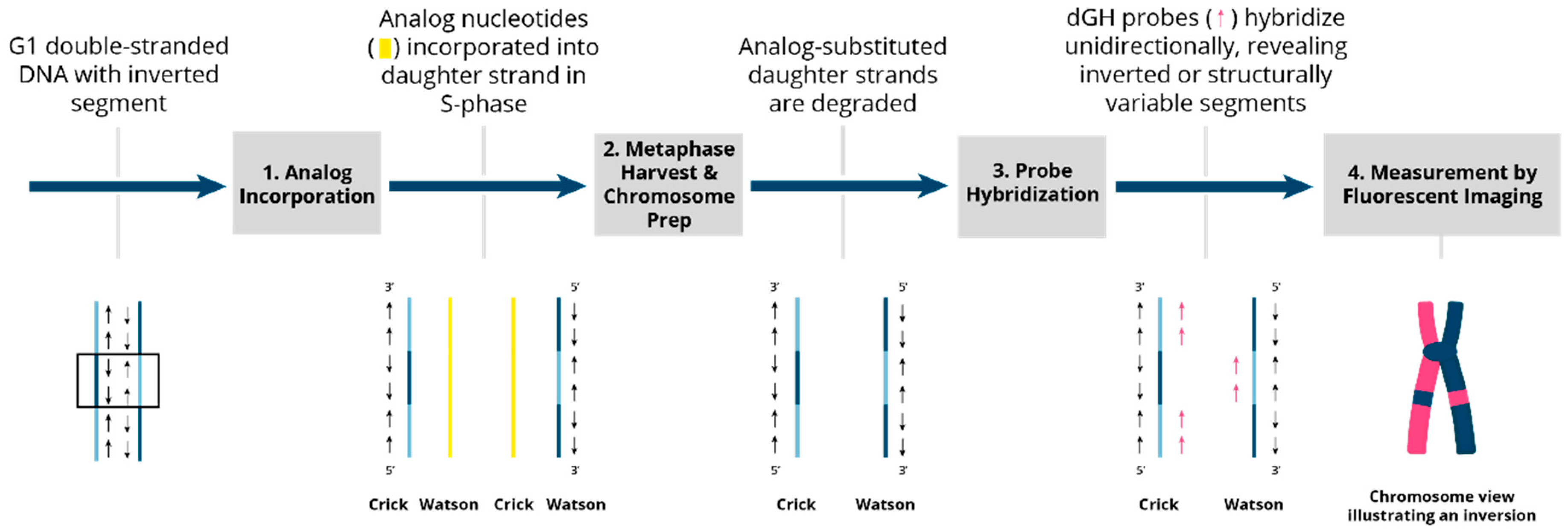
3.1. Directional Genomic Hybridization (dGH) Provides a Direct and Genome-Wide Visualization of Structural Variants
3.2. Whole-Genome Discovery vs. Targeted Detection Using dGH
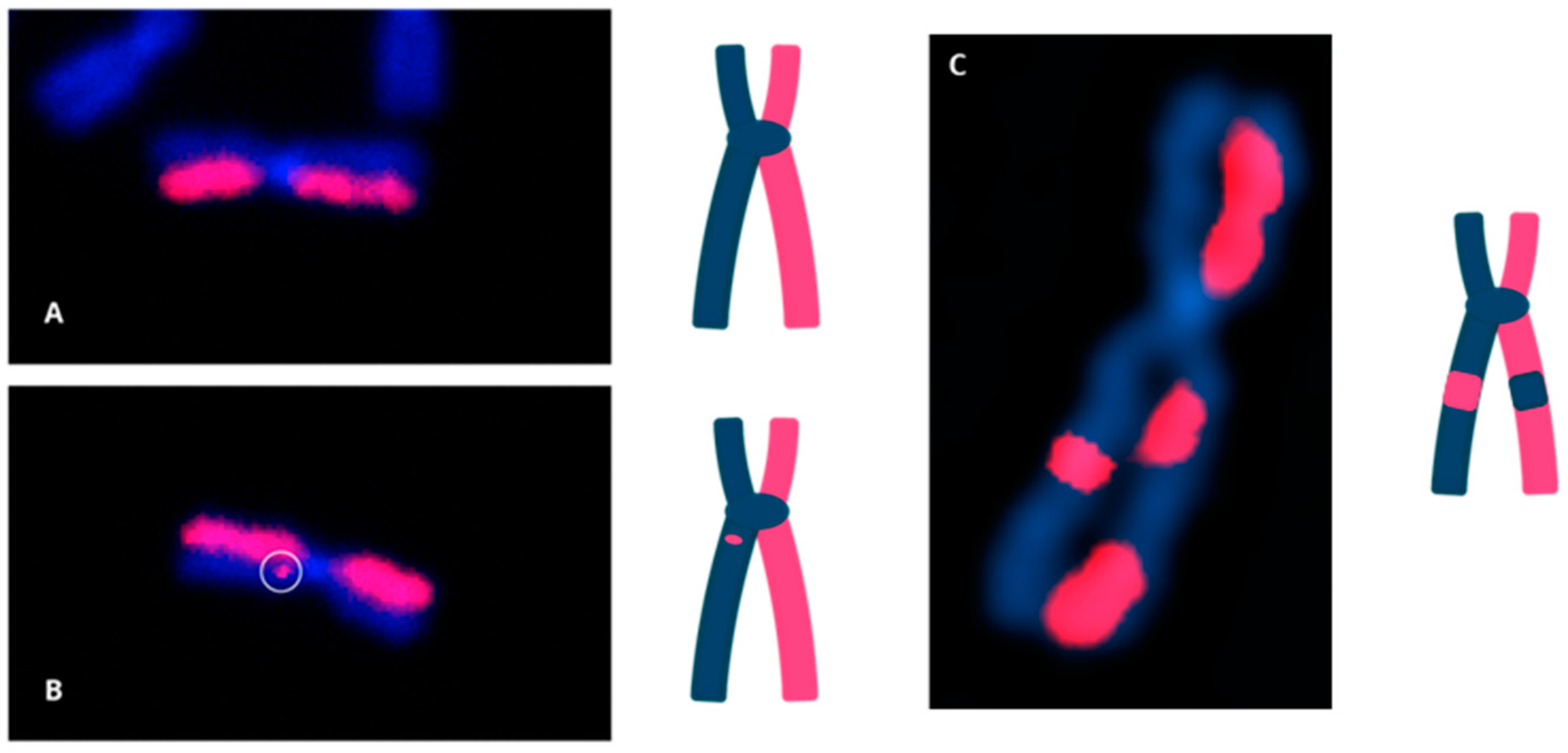
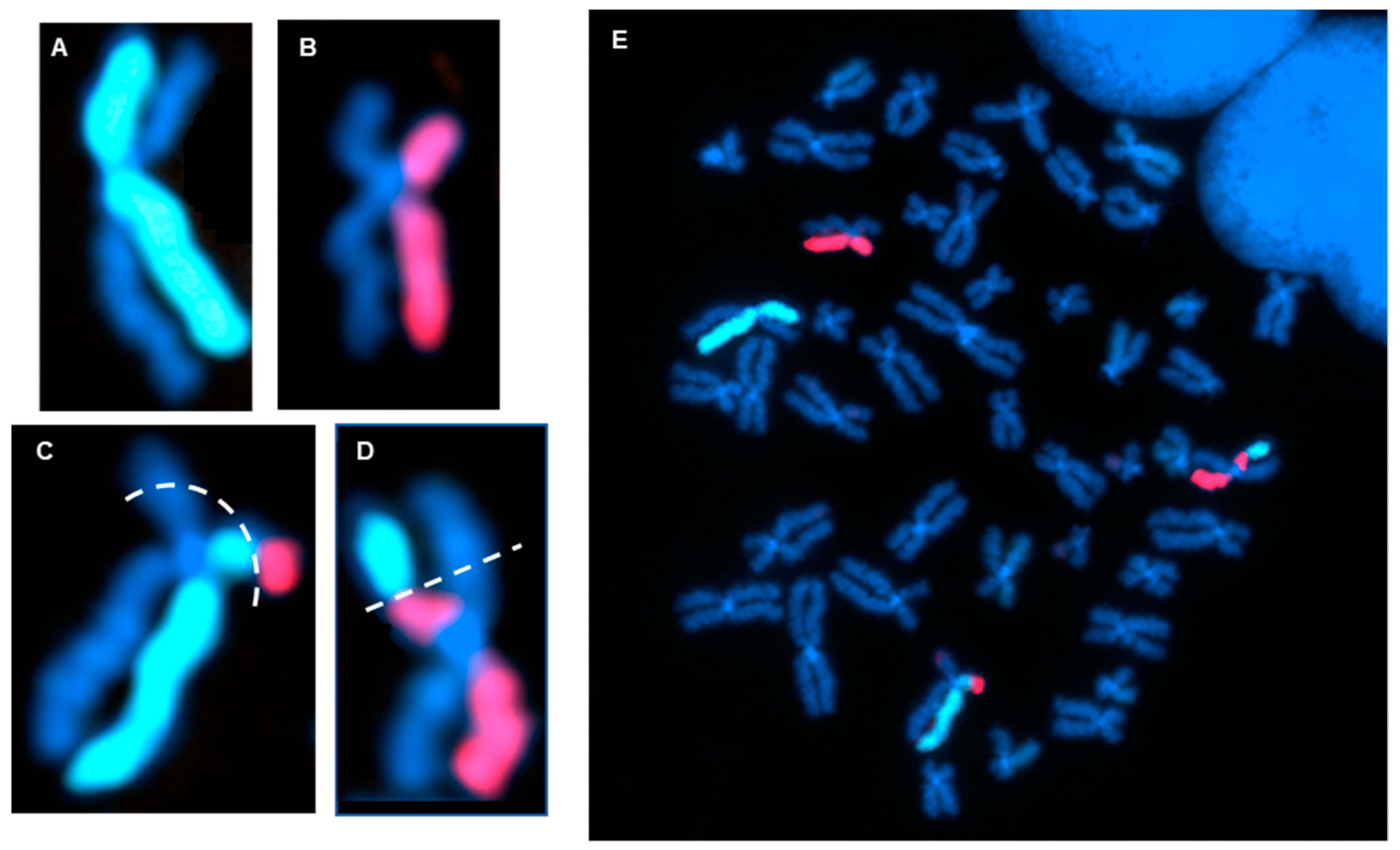
3.3. Measurement of the Products of Mis-Repair in Batches of Edited Cells
- −
- Reciprocal translocations between the edit site on non-homologous chromosomes.
- −
- Inversions between edit sites on the target chromosome and a different site on the same chromosome, as well as translocations between edit sites on the target chromosome and various sites on different chromosomes.
- −
- Complex variants that defied simple naming convention definitions.
- −
- Chromothripsis products and micronuclei.
4. Discussion
4.1. Classical Cytogenetics Techniques Do Not Provide a Comprehensive Assessment of Potential Structural Outcomes of Gene Editing
4.2. Assessment of Genomic Structural Variation Using dGH Complements Bioinformatic Sequencing Techniques Used to Predict and Measure Classical Off-Target Effects Associated with Gene Editing
4.3. Indirect Detection of Structural Variants through Fusion Gene Products
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef]
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef]
- Hunt, J.M.T.; Samson, C.A.; Rand, A.D.; Sheppard, H.M. Unintended CRISPR-Cas9 editing outcomes: A review of the detection and prevalence of structural variants generated by gene-editing in human cells. Hum. Genet. 2023, 142, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Bailey, S.M.; Bedford, J.S. Studies on chromosome aberration induction: What can they tell us about DNA repair? DNA Repair 2006, 5, 1171–1181. [Google Scholar] [CrossRef]
- Cornforth, M.N.; Bedford, J.S.; Bailey, S.M. Destabilizing Effects of Ionizing Radiation on Chromosomes: Sizing up the Damage. Cytogenet. Genome Res. 2021, 161, 328–351. [Google Scholar] [CrossRef]
- Hoijer, I.; Emmanouilidou, A.; Ostlund, R.; van Schendel, R.; Bozorgpana, S.; Tijsterman, M.; Feuk, L.; Gyllensten, U.; den Hoed, M.; Ameur, A. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun. 2022, 13, 627. [Google Scholar] [CrossRef]
- van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Lobrich, M. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem. J. 2015, 471, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, M.N.; Bedford, J.S. Ionizing radiation damage and its early development in chromosomes. Adv. Radiat. Biol. 1993, 17, 423–496. [Google Scholar] [CrossRef]
- Ray, F.A.; Robinson, E.; McKenna, M.; Hada, M.; George, K.; Cucinotta, F.; Goodwin, E.H.; Bedford, J.S.; Bailey, S.M.; Cornforth, M.N. Directional genomic hybridization: Inversions as a potential biodosimeter for retrospective radiation exposure. Radiat. Environ. Biophys. 2014, 53, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ray, F.A.; Zimmerman, E.; Robinson, B.; Cornforth, M.N.; Bedford, J.S.; Goodwin, E.H.; Bailey, S.M. Directional genomic hybridization for chromosomal inversion discovery and detection. Chromosome Res. 2013, 21, 165–174. [Google Scholar] [CrossRef]
- McKenna, M.J.; Robinson, E.; Taylor, L.; Tompkins, C.; Cornforth, M.N.; Simon, S.L.; Bailey, S.M. Chromosome Translocations, Inversions and Telomere Length for Retrospective Biodosimetry on Exposed U.S. Atomic Veterans. Radiat. Res. 2019, 191, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, M.N.; Anur, P.; Wang, N.; Robinson, E.; Ray, F.A.; Bedford, J.S.; Loucas, B.D.; Williams, E.S.; Peto, M.; Spellman, P.; et al. Molecular Cytogenetics Guides Massively Parallel Sequencing of a Radiation-Induced Chromosome Translocation in Human Cells. Radiat. Res. 2018, 190, 88–97. [Google Scholar] [CrossRef]
- Savage, J.R. Classification and relationships of induced chromosomal structual changes. J. Med. Genet. 1976, 13, 103–122. [Google Scholar] [CrossRef]
- Bender, M.A.; Griggs, H.G.; Bedford, J.S. Mechanisms of chromosomal aberration production. 3. Chemicals and ionizing radiation. Mutat. Res. 1974, 23, 197–212. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef]
- Li, W.; Li, F.; Huang, Q.; Shen, J.; Wolf, F.; He, Y.; Liu, X.; Hu, Y.A.; Bedford, J.S.; Li, C.Y. Quantitative, noninvasive imaging of radiation-induced DNA double-strand breaks in vivo. Cancer Res. 2011, 71, 4130–4137. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.A.; Nagasawa, H.; Weil, M.M.; Genik, P.C.; Little, J.B.; Bedford, J.S. gamma-H2AX foci after low-dose-rate irradiation reveal atm haploinsufficiency in mice. Radiat. Res. 2006, 166, 47–54. [Google Scholar] [CrossRef]
- Kato, T.A.; Nagasawa, H.; Weil, M.M.; Little, J.B.; Bedford, J.S. Levels of gamma-H2AX Foci after low-dose-rate irradiation reveal a DNA DSB rejoining defect in cells from human ATM heterozygotes in two at families and in another apparently normal individual. Radiat. Res. 2006, 166, 443–453. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed]
- Sansbury, B.M.; Hewes, A.M.; Kmiec, E.B. Understanding the diversity of genetic outcomes from CRISPR-Cas generated homology-directed repair. Commun. Biol. 2019, 2, 458. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Goedecke, W.; Obe, G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 2000, 15, 289–302. [Google Scholar] [CrossRef]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2018, 809, 81–87. [Google Scholar] [CrossRef]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef]
- Helmink, B.A.; Sleckman, B.P. The response to and repair of RAG-mediated DNA double-strand breaks. Annu. Rev. Immunol. 2012, 30, 175–202. [Google Scholar] [CrossRef]
- Poot, M.; Haaf, T. Mechanisms of Origin, Phenotypic Effects and Diagnostic Implications of Complex Chromosome Rearrangements. Mol. Syndr. 2015, 6, 110–134. [Google Scholar] [CrossRef]
- Hinz, J.M.; Tebbs, R.S.; Wilson, P.F.; Nham, P.B.; Salazar, E.P.; Nagasawa, H.; Urbin, S.S.; Bedford, J.S.; Thompson, L.H. Repression of mutagenesis by Rad51D-mediated homologous recombination. Nucleic Acids Res. 2006, 34, 1358–1368. [Google Scholar] [CrossRef]
- Richardson, C.; Moynahan, M.E.; Jasin, M. Double-strand break repair by interchromosomal recombination: Suppression of chromosomal translocations. Genes. Dev. 1998, 12, 3831–3842. [Google Scholar] [CrossRef]
- Cornforth, M.N.; Bedford, J.S. X-ray—Induced breakage and rejoining of human interphase chromosomes. Science 1983, 222, 1141–1143. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Lomova, A.; Clark, D.N.; Campo-Fernandez, B.; Flores-Bjurstrom, C.; Kaufman, M.L.; Fitz-Gibbon, S.; Wang, X.; Miyahira, E.Y.; Brown, D.; DeWitt, M.A.; et al. Improving Gene Editing Outcomes in Human Hematopoietic Stem and Progenitor Cells by Temporal Control of DNA Repair. Stem Cells 2019, 37, 284–294. [Google Scholar] [CrossRef]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 2011, 20, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Poot, M. Genes, Proteins, and Biological Pathways Preventing Chromothripsis. Methods Mol. Biol. 2018, 1769, 231–251. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Coebergh van den Braak, R.R.J.; Pieterse, M.; van Roosmalen, M.J.; Sieuwerts, A.M.; Stangl, C.; Brunekreef, R.; Lalmahomed, Z.S.; Ooft, S.; van Galen, A.; et al. A Systematic Analysis of Oncogenic Gene Fusions in Primary Colon Cancer. Cancer Res. 2017, 77, 3814–3822. [Google Scholar] [CrossRef]
- Stangl, C.; de Blank, S.; Renkens, I.; Westera, L.; Verbeek, T.; Valle-Inclan, J.E.; Gonzalez, R.C.; Henssen, A.G.; van Roosmalen, M.J.; Stam, R.W.; et al. Partner independent fusion gene detection by multiplexed CRISPR-Cas9 enrichment and long read nanopore sequencing. Nat. Commun. 2020, 11, 2861. [Google Scholar] [CrossRef]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; Van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational signatures of ionizing radiation in second malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef]
- Rayner, E.; Durin, M.A.; Thomas, R.; Moralli, D.; O’Cathail, S.M.; Tomlinson, I.; Green, C.M.; Lewis, A. CRISPR-Cas9 Causes Chromosomal Instability and Rearrangements in Cancer Cell Lines, Detectable by Cytogenetic Methods. CRISPR J. 2019, 2, 406–416. [Google Scholar] [CrossRef]
- Skryabin, B.V.; Kummerfeld, D.M.; Gubar, L.; Seeger, B.; Kaiser, H.; Stegemann, A.; Roth, J.; Meuth, S.G.; Pavenstadt, H.; Sherwood, J.; et al. Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9-mediated genome editing events. Sci. Adv. 2020, 6, eaax2941. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.E6. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef] [PubMed]
- Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; Reeves, C.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef]
- Zhao, X.; Collins, R.L.; Lee, W.P.; Weber, A.M.; Jun, Y.; Zhu, Q.; Weisburd, B.; Huang, Y.; Audano, P.A.; Wang, H.; et al. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am. J. Hum. Genet. 2021, 108, 919–928. [Google Scholar] [CrossRef]
- De Pascalis, I.; Pilato, B.; Mazzotta, A.; Dell’Endice, T.S.; Rubini, V.; Simone, G.; Paradiso, A.; Aiello, V.; Mangia, A. Sister chromatid exchange: A possible approach to characterize familial breast cancer patients. Oncol. Rep. 2015, 33, 930–934. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Thompson, L.H. Molecular mechanisms of sister-chromatid exchange. Mutat. Res. 2007, 616, 11–23. [Google Scholar] [CrossRef]
- Kadhim, M.A.; Macdonald, D.A.; Goodhead, D.T.; Lorimore, S.A.; Marsden, S.J.; Wright, E.G. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature 1992, 355, 738–740. [Google Scholar] [CrossRef]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef]
- Robinson, E.; McKenna, M.J.; Bedford, J.S.; Goodwin, E.H.; Cornforth, M.N.; Bailey, S.M.; Ray, F.A. Directional Genomic Hybridization (dGH) for Detection of Intrachromosomal Rearrangements. Methods Mol. Biol. 2019, 1984, 107–116. [Google Scholar] [CrossRef]
- McKenna, M.J.; Robinson, E.; Goodwin, E.H.; Cornforth, M.N.; Bailey, S.M. Telomeres and NextGen CO-FISH: Directional Genomic Hybridization (Telo-dGH). Methods Mol. Biol. 2017, 1587, 103–112. [Google Scholar] [CrossRef]
- Simon, S.L.; Bailey, S.M.; Beck, H.L.; Boice, J.D.; Bouville, A.; Brill, A.B.; Cornforth, M.N.; Inskip, P.D.; McKenna, M.J.; Mumma, M.T.; et al. Estimation of Radiation Doses to U.S. Military Test Participants from Nuclear Testing: A Comparison of Historical Film-Badge Measurements, Dose Reconstruction and Retrospective Biodosimetry. Radiat. Res. 2019, 191, 297–310. [Google Scholar] [CrossRef]
- Luxton, J.J.; McKenna, M.J.; Lewis, A.; Taylor, L.E.; George, K.A.; Dixit, S.M.; Moniz, M.; Benegas, W.; Mackay, M.J.; Mozsary, C.; et al. Telomere Length Dynamics and DNA Damage Responses Associated with Long-Duration Spaceflight. Cell Rep. 2020, 33, 108457. [Google Scholar] [CrossRef]
- Luxton, J.J.; McKenna, M.J.; Lewis, A.M.; Taylor, L.E.; Jhavar, S.G.; Swanson, G.P.; Bailey, S.M. Telomere Length Dynamics and Chromosomal Instability for Predicting Individual Radiosensitivity and Risk via Machine Learning. J. Pers. Med. 2021, 11, 188. [Google Scholar] [CrossRef]
- McKenna, M.J.; Bailey, S.M. Chromosomal and Telomeric biomarkers of normal tissue injury to evaluate risk of degenerative health effects (secondary malignancy, cardiovascular disease) post radiation therapy. Transl. Cancer Res. 2017, 6, S789–S794. [Google Scholar] [CrossRef]
- Bailey, S.M.; Goodwin, E.H.; Cornforth, M.N. Strand-specific fluorescence in situ hybridization: The CO-FISH family. Cytogenet. Genome Res. 2004, 107, 14–17. [Google Scholar] [CrossRef]
- Sutherland, G.R.; Baker, E.; Richards, R.I. Fragile sites still breaking. Trends Genet. 1998, 14, 501–506. [Google Scholar] [CrossRef]
- Wahls, W.P.; Wallace, L.J.; Moore, P.D. Hypervariable minisatellite DNA is a hotspot for homologous recombination in human cells. Cell 1990, 60, 95–103. [Google Scholar] [CrossRef]
- Debrauwere, H.; Buard, J.; Tessier, J.; Aubert, D.; Vergnaud, G.; Nicolas, A. Meiotic instability of human minisatellite CEB1 in yeast requires DNA double-strand breaks. Nat. Genet. 1999, 23, 367–371. [Google Scholar] [CrossRef]
- Haber, J.E. DNA recombination: The replication connection. Trends Biochem. Sci. 1999, 24, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Daser, A.; Dear, P.; Wood, H.; Rabbitts, P.; Rabbitts, T. Nonreciprocal chromosomal translocations in renal cancer involve multiple DSBs and NHEJ associated with breakpoint inversion but not necessarily with transcription. Genes. Chromosomes Cancer 2013, 52, 402–409. [Google Scholar] [CrossRef]
- Cornforth, M.N. Analyzing radiation-induced complex chromosome rearrangements by combinatorial painting. Radiat. Res. 2001, 155, 643–659. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, M.N.; Bedford, J.S. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat. Res. 1987, 111, 385–405. [Google Scholar] [CrossRef]
- Forster, J.C.; Douglass, M.J.J.; Phillips, W.M.; Bezak, E. Stochastic multicellular modeling of x-ray irradiation, DNA damage induction, DNA free-end misrejoining and cell death. Sci. Rep. 2019, 9, 18888. [Google Scholar] [CrossRef] [PubMed]
- Sakofsky, C.J.; Saini, N.; Klimczak, L.J.; Chan, K.; Malc, E.P.; Mieczkowski, P.A.; Burkholder, A.B.; Fargo, D.; Gordenin, D.A. Repair of multiple simultaneous double-strand breaks causes bursts of genome-wide clustered hypermutation. PLoS Biol. 2019, 17, e3000464. [Google Scholar] [CrossRef] [PubMed]
- Shuryak, I.; Loucas, B.D.; Cornforth, M.N. Straightening Beta: Overdispersion of Lethal Chromosome Aberrations following Radiotherapeutic Doses Leads to Terminal Linearity in the Alpha-Beta Model. Front. Oncol. 2017, 7, 318. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.A.; Okayasu, R.; Bedford, J.S. Signatures of DNA double strand breaks produced in irradiated G1 and G2 cells persist into mitosis. J. Cell Physiol. 2009, 219, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, L.; Sachs, R.K.; Vazquez, M.; Cornforth, M.N. Radiation-induced chromosome aberrations: Insights gained from biophysical modeling. Bioessays 2002, 24, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Adikusuma, F.; Piltz, S.; Corbett, M.A.; Turvey, M.; McColl, S.R.; Helbig, K.J.; Beard, M.R.; Hughes, J.; Pomerantz, R.T.; Thomas, P.Q. Large deletions induced by Cas9 cleavage. Nature 2018, 560, E8–E9. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Puig, M.; Casillas, S.; Villatoro, S.; Caceres, M. Human inversions and their functional consequences. Brief. Funct. Genom. 2015, 14, 369–379. [Google Scholar] [CrossRef]
- Fan, Y.S.; Siu, V.M.; Jung, J.H.; Xu, J. Sensitivity of multiple color spectral karyotyping in detecting small interchromosomal rearrangements. Genet. Test. 2000, 4, 9–14. [Google Scholar] [CrossRef]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Giannoukos, G.; Ciulla, D.M.; Marco, E.; Abdulkerim, H.S.; Barrera, L.A.; Bothmer, A.; Dhanapal, V.; Gloskowski, S.W.; Jayaram, H.; Maeder, M.L.; et al. UDiTaS, a genome editing detection method for indels and genome rearrangements. BMC Genom. 2018, 19, 212. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Gobet, N.; Cruz-Davalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019, 20, 246. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.J.; Borrow, J.; Goddard, A.D. Chromosome aberrations and cancer. Science 1991, 254, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Demura, M.; Martin, R.M.; Shozu, M.; Sebastian, S.; Takayama, K.; Hsu, W.T.; Schultz, R.A.; Neely, K.; Bryant, M.; Mendonca, B.B.; et al. Regional rearrangements in chromosome 15q21 cause formation of cryptic promoters for the CYP19 (aromatase) gene. Hum. Mol. Genet. 2007, 16, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Rodig, S.J.; Chirieac, L.R.; Janne, P.A. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur. J. Cancer 2010, 46, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ichikawa, M.; Kamikubo, Y.; Kurokawa, M. Acute myeloid leukemia with cryptic CBFB-MYH11 type D. Int. J. Clin. Exp. Pathol. 2013, 6, 110–112. [Google Scholar] [PubMed]
- Monma, F.; Nishii, K.; Shiga, J.; Sugahara, H.; Lorenzo, F.t.; Watanabe, Y.; Kawakami, K.; Hosokai, N.; Yamamori, S.; Katayama, N.; et al. Detection of the CBFB/MYH11 fusion gene in de novo acute myeloid leukemia (AML): A single-institution study of 224 Japanese AML patients. Leuk. Res. 2007, 31, 471–476. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Stringer, J.R.; Blough, R.; Medvedovic, M.; Fagin, J.A.; Nikiforov, Y.E. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 2000, 290, 138–141. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Lee, J.J.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kuzminov, A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 1999, 63, 751–813. [Google Scholar] [CrossRef]
- Cortes-Ledesma, F.; Aguilera, A. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep. 2006, 7, 919–926. [Google Scholar] [CrossRef]
- OECD. Test No. 473: In Vitro Mammalian Chromosomal Aberration Test; OECD: Paris, France, 2016. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Purpose | Details |
|---|---|---|
Chromatid paint dGH (SCREEN) | Full coverage of single sister chromatid on target chromosome Can be used to target chromosomes of interest or in a whole-genome format | Discovery of inversions or other aberrations, such as translocations, where location, type, instance, and/or prevalence are unknown Lower limit of detection: >2 KB Detection of off-target effects in edited cell populations |
Targeted dGH (In-site) | Targeted probe designed for an area of interest | Useful for studying a specific gene/target (i.e., edit site or known disease target) Detection of unintended on-target effects in edited cell populations |
| Method | Purpose | Strengths | Limitations |
|---|---|---|---|
| Giemsa staining/G-banding | Staining of AT-rich regions to create a unique banding pattern for each chromosome Generation of karyograms International standard reference for gene mapping | Identification of large-scale chromosomal aberrations | Low resolution (10 Mb) Cannot detect small inversions or those that do not grossly alter the banding pattern Lack of targeted information |
| Fluorescence in situ hybridization (FISH) | Fluorescence probe-based detection of target chromosomes. Can be against a specific target region within a chromosome or an entire chromosome (“whole chromosome painting”) | Can detect targeted, specific structures and rearrangements | Lack of commercially available, validated probes for many species and targets Widely varied procedures Cannot directly detect inversions Targeted analysis only |
| Spectral karyotyping (SKY) or multiplex or multifluor combinatorial FISH (mFISH) | Evolution of FISH that allows for visualization and unique identification of all painted chromosomes in a single hybridization reaction | Simultaneous visualization of all chromosomes Can detect interchange chromosomal rearrangements | Estimated resolution of 0.5–2 Mb for interchromosomal rearrangements [88] Cannot detect inversions or non-lethal deletions |
| Comparative genomic hybridization (CGH) | Identification of copy number variation | Detection of deletions and amplifications | Cannot detect inversions or translocations |
| Genomic Vision- Genomic Morse Code (GMC) | High-resolution banding of a genomic region of interest | Detection of structural variants and copy number variants Visualization of hard to sequence regions | Lack of whole-genome information (can visualize up to several Mb) Pooled, double-stranded DNA does not provide data on a cell-by-cell basis. No inversion detection |
| BioNano-Genome Mapping | Genome mapping using a sequence of probes to generate sequence-based fluorescent patterns | Detection of variants, insertions, and deletions | Pooled, double-stranded DNA does not provide data on a cell-by-cell basis. No inversion detection |
| Method | Strengths | Limitations |
|---|---|---|
| Targeted sequencing | Specific data about a genomic region of interest | Lack of whole-genome information |
| Paired-end sequencing | Detection of structural variants based on end pairs that are mapped abnormally far apart on the normal genome sequence | Cannot reliably detect variants in repetitive regions; heterozygous/rare variants |
| Long-read sequencing | Longer reads allow for improved detection of structural variants in repetitive regions | Cannot reliably detect heterozygous/rare variants High error rate |
| Single-molecule real-time sequencing (SMRT) | Single DNA template is sequenced with a single DNA polymerase Evolution of long-read sequencing | Cannot reliably detect rare variants due to pooled cell samples Lower throughput due to high cost and time requirements |
| Unidirectional sequencing | Targeted primer used with bridge adaptors on sheared DNA Often designed for detection of off-target DSBs associated with CRISPR/Cas9 | Requires specialized equipment for the shearing of DNA Cannot reliably detect inversions |
| Single-cell sequencing | Isolation and lysis of single-cell preparations allow for analysis on a cell-by-cell basis rather than population-based information | High level of noise can confound results Low capture efficiency during single-cell isolation Low throughput |
| 10X Genomics Next-GEM sequencing | Combines the specificity of short-read sequencing with the broad range of information captured by long-read sequencing Can detect large structural variants and provide sequence-specific information for single cells | Bioinformatic calculation of structure rather than direct detection makes structural variant detection difficult Susceptible to selection bias |
| Method | Purpose | Strengths | Limitations |
|---|---|---|---|
| NanoString nCounter | Multiplexed gene expression and biomarker analysis platform that uses fluorescent barcodes for identification of sequences | Multiplexing of regions for analysis of copy number variants Fusion gene detection | Targeted approach; lack of whole-genome information (up to 800 targets) Does not provide data on a cell-by-cell basis |
| Anchored Multiplex PCR (ArcherDX) | NGS-based targeted approach for detection of genetic variants | Discovery of fusion gene partners Detection of splice variants | Targeted NGS approach (lack of whole-genome off-target effects) Does not provide data on a cell-by-cell basis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, S.M.; Cross, E.M.; Kinner-Bibeau, L.; Sebesta, H.C.; Bedford, J.S.; Tompkins, C.J. Monitoring Genomic Structural Rearrangements Resulting from Gene Editing. J. Pers. Med. 2024, 14, 110. https://doi.org/10.3390/jpm14010110
Bailey SM, Cross EM, Kinner-Bibeau L, Sebesta HC, Bedford JS, Tompkins CJ. Monitoring Genomic Structural Rearrangements Resulting from Gene Editing. Journal of Personalized Medicine. 2024; 14(1):110. https://doi.org/10.3390/jpm14010110
Chicago/Turabian StyleBailey, Susan M., Erin M. Cross, Lauren Kinner-Bibeau, Henry C. Sebesta, Joel S. Bedford, and Christopher J. Tompkins. 2024. "Monitoring Genomic Structural Rearrangements Resulting from Gene Editing" Journal of Personalized Medicine 14, no. 1: 110. https://doi.org/10.3390/jpm14010110
APA StyleBailey, S. M., Cross, E. M., Kinner-Bibeau, L., Sebesta, H. C., Bedford, J. S., & Tompkins, C. J. (2024). Monitoring Genomic Structural Rearrangements Resulting from Gene Editing. Journal of Personalized Medicine, 14(1), 110. https://doi.org/10.3390/jpm14010110