
High-Salt Diet Exacerbates H. pylori Infection and Increases Gastric Cancer Risks

 , , and
, , and 
Abstract
1. Introduction
1.1. Genetics
1.1.1. CagA
1.1.2. IceA
1.1.3. BabA
1.2. Clinical Presentation
2. Materials and Methods
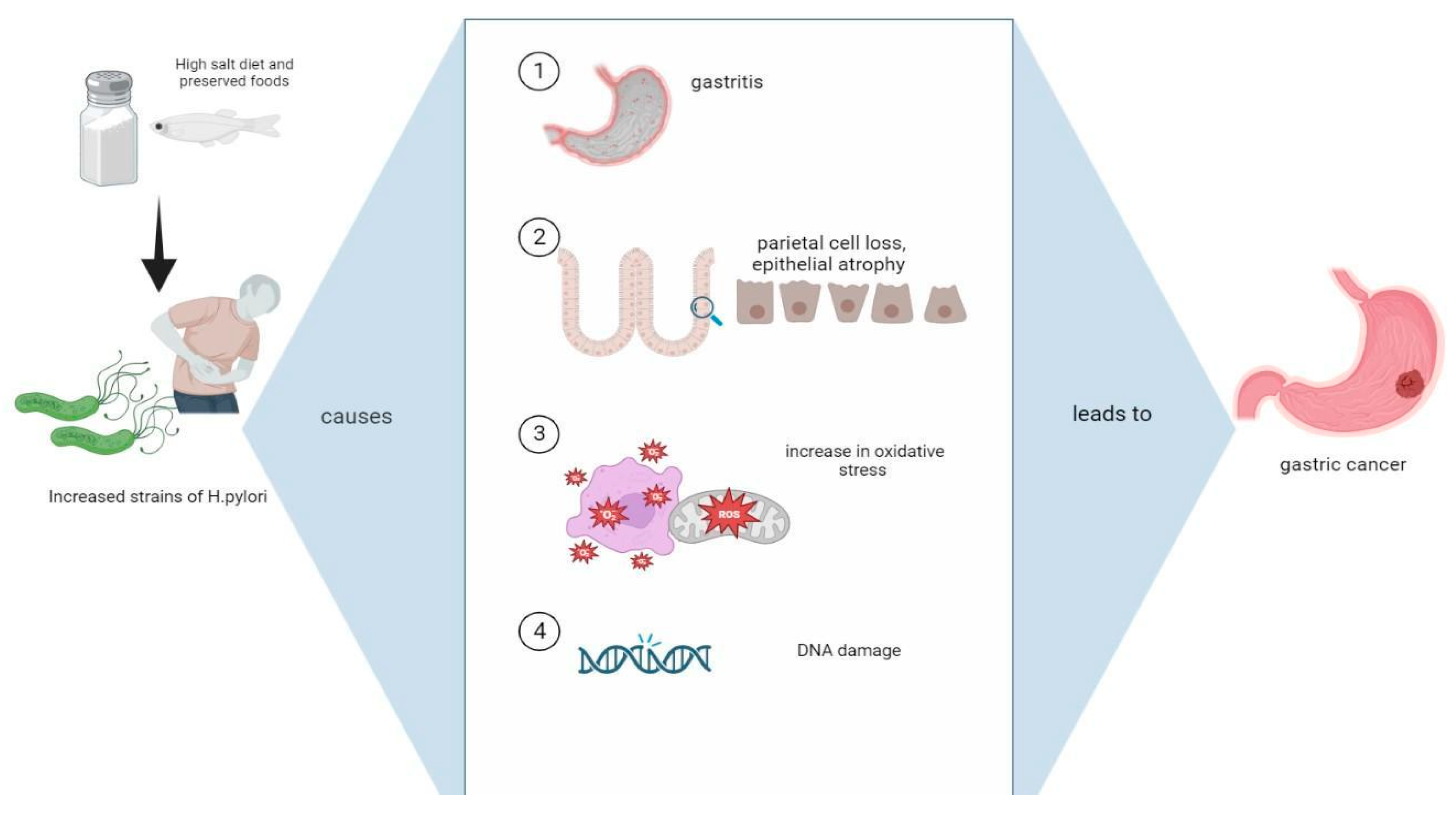
3. Mechanisms Causing Gastric Cancer from the Synergistic Influence of a High-Salt Diet with H. pylori Infection
3.1. Damage to Mucosal Barrier and Intestinal Metaplasia
3.2. Foods High in Nitrites
3.3. Alterations in Composition of H. pylori Strains
3.4. Oxidative Stress
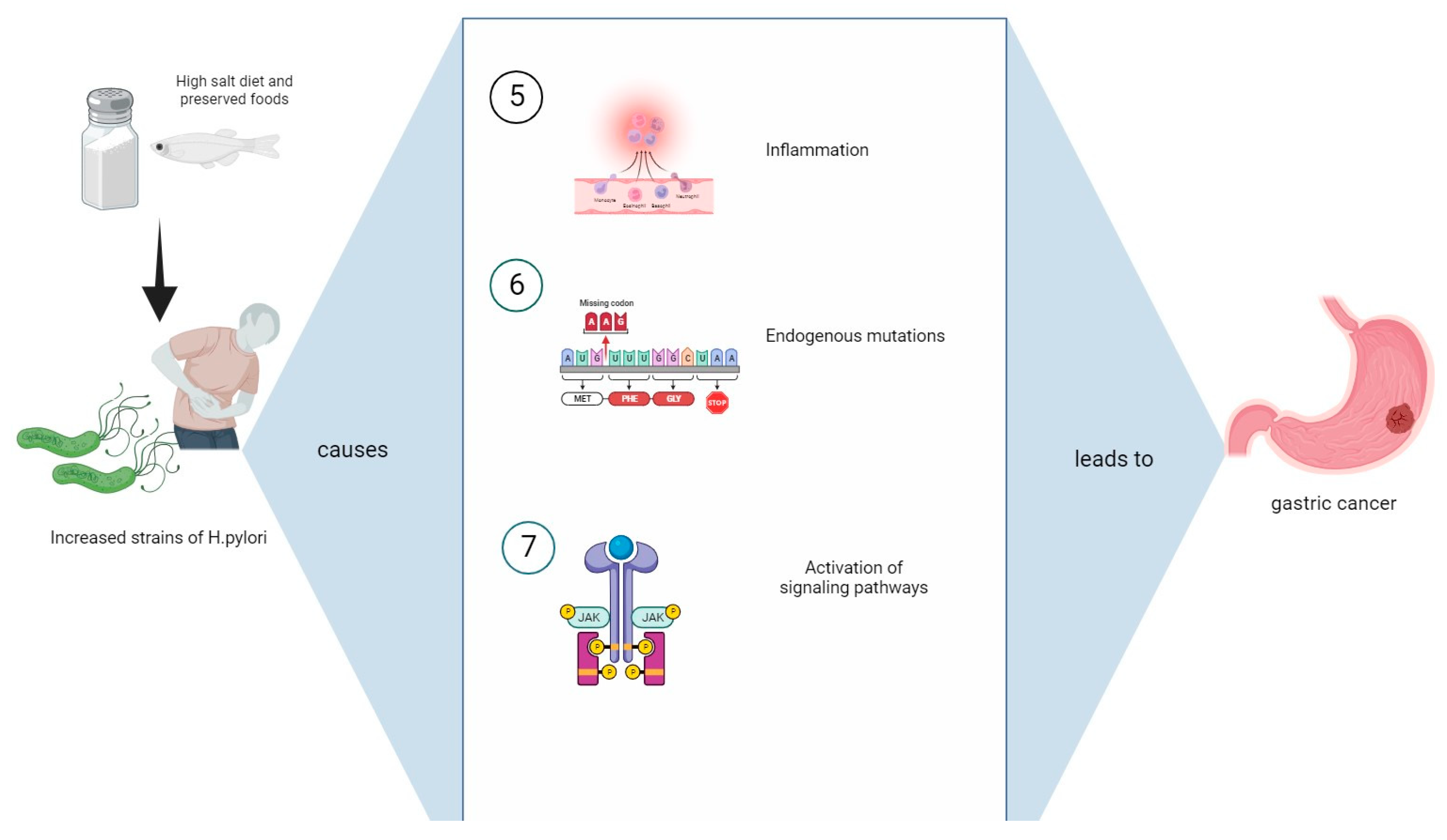
3.5. Endogenous Mutations

{kind=link}
{kind=link}
| Inflammation Effects of an HSD with H. pylori Infection | Mechanisms Leading to Gastric Cancer | References |
|---|---|---|
| Mucosal barrier damage | Increased mitotic cell division and turnover | [32,33,37,38,39,40] |
| Induction of intestinal hyperplasia and metaplasia | [38,39] | |
| Cellular damage | Irreversible lipid peroxidation, organelle swelling, and mitochondrial dysfunction | [40] |
| Alteration of H. pylori gene expression | Increased expression of CagA, VacA, BabA, and SabA | [14,32,50,58,59,79,80,84,85,86,87,88,89,90,91] |
| Oxidative stress via ROSs | Apoptosis of gastric cells via the production of superoxide anions and hydrogen peroxide | [68,71,75,76] |
| Inflammation | Activation of TNFα, interleukins, COX-2, and Th17 | [19,55,64,77,78,92,93,94,95,96,97,98] |
3.6. Management of Helicobacter Pylori Infection: Antibiotics, Probiotics, Natural Treatments, and Future Perspectives
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020–2040: A population-based modelling study. eClinicalMedicine 2022, 47, 101404. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Shan, F.; Ying, X.; Li, Z. Global burden prediction of gastric cancer during demographic transition from 2020 to 2040. Chin. Med. J. 2023, 136, 397–406. [Google Scholar] [CrossRef]
- Fock, K.M. Review article: The epidemiology and prevention of gastric cancer. Aliment. Pharmacol. Ther. 2014, 40, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Asombang, A.W.; Ibdah, J.A. Characteristics of gastric cancer in Asia. World J. Gastroenterol. 2014, 20, 4483–4490. [Google Scholar] [CrossRef] [PubMed]
- Raei, N.; Behrouz, B.; Zahri, S.; Latifi-Navid, S. Helicobacter pylori Infection and Dietary Factors Act Synergistically to Promote Gastric Cancer. Asian Pac. J. Cancer Prev. APJCP 2016, 17, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Krzyżek, P.; Gościniak, G. Morphology of Helicobacter pylori as a result of peptidoglycan and cytoskeleton rearrangements. Prz. Gastroenterol. 2018, 13, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, M.; Reddy, K.M.; Marsicano, E. Peptic Ulcer Disease and Helicobacter pylori infection. Mo. Med. 2018, 115, 219–224. [Google Scholar]
- Suerbaum, S.; Smith, J.M.; Bapumia, K.; Morelli, G.; Smith, N.H.; Kunstmann, E.; Dyrek, I.; Achtman, M. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA 1998, 95, 12619–12624. [Google Scholar] [CrossRef]
- Suerbaum, S.; Josenhans, C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007, 5, 441–452. [Google Scholar] [CrossRef]
- García-Ortíz, M.V.; Marsin, S.; Arana, M.E.; Gasparutto, D.; Guérois, R.; Kunkel, T.A.; Radicella, J.P. Unexpected role for Helicobacter pylori DNA polymerase I as a source of genetic variability. PLoS Genet. 2011, 7, e1002152. [Google Scholar] [CrossRef]
- Varga, M.G.; Shaffer, C.L.; Sierra, J.C.; Suarez, G.; Piazuelo, M.B.; Whitaker, M.E.; Gallo, J.R.; Krishna, U.S.; Delgado, A.; Gomez, M.A.; et al. Pathogenic Helicobacter pylori strains translocate DNA and activate TLR9 via the cancer-associated cag Type IV secretion system. Oncogene 2016, 1, 6262–6269. [Google Scholar] [CrossRef] [PubMed]
- Dorer, M.S.; Sessler, T.H.; Salama, N.R. Recombination and DNA repair in Helicobacter pylori. Annu. Rev. Microbiol. 2011, 65, 329–348. [Google Scholar] [CrossRef]
- Roesler, B.M.; Rabelo-Gonçalves, E.M.; Zeitune, J.M. Virulence Factors of Helicobacter pylori: A Review. Clin. Med. Insights Gastroenterol. 2014, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Moss, S.F. Helicobacter pylori virulence factors in gastric carcinogenesis. Cancer Lett. 2009, 282, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Caston, R.R.; Sierra, J.C.; Foegeding, N.J.; Truelock, M.D.; Campbell, A.M.; Frick-Cheng, A.E.; Bimczok, D.; Wilson, K.T.; McClain, M.S.; Cover, T.L. Functional Properties of Helicobacter pylori VacA Toxin m1 and m2 Variants. Infect. Immun. 2020, 88, e00032-20. [Google Scholar] [CrossRef] [PubMed]
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. CagA Phosphorylation in Helicobacter pylori-Infected B Cells Is Mediated by the Nonreceptor Tyrosine Kinases of the Src and Abl Families. Infect. Immun. 2016, 84, 2671–2680. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Yamaoka, Y. Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity. Int. J. Mol. Sci. 2020, 21, 7430. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Kodama, T.; Gutierrez, O.; Kim, J.G.; Kashima, K.; Graham, D.Y. Relationship between Helicobacter pylori iceA, cagA, and vacA status and clinical outcome: Studies in four different countries. J. Clin. Microbiol. 1999, 37, 2274–2279. [Google Scholar] [CrossRef]
- Hage, N.; Howard, T.; Phillips, C.; Brassington, C.; Overman, R.; Debreczeni, J.; Gellert, P.; Stolnik, S.; Winkler, G.S.; Falcone, F.H. Structural basis of Lewis(b) antigen binding by the Helicobacter pylori adhesin BabA. Sci. Adv. 2015, 1, e1500315. [Google Scholar] [CrossRef]
- Xu, C.; Soyfoo, D.M.; Wu, Y.; Xu, S. Virulence of Helicobacter pylori outer membrane proteins: An updated review. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2020, 39, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Odenbreit, S.; Swoboda, K.; Barwig, I.; Ruhl, S.; Borén, T.; Koletzko, S.; Haas, R. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect. Immun. 2009, 77, 3782–3790. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Vahedi, A.; Maghdouri, Z.; Shokri-Shirvani, J. Role of food in environmental transmission of Helicobacter pylori. Casp. J. Intern. Med. 2017, 8, 146–152. [Google Scholar] [CrossRef]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Sasazuki, S.; Kobayashi, M.; Sasaki, S. Salt and salted food intake and subsequent risk of gastric cancer among middle-aged Japanese men and women. Br. J. Cancer 2004, 90, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.R.; He, F.J.; MacGregor, G.A.; Graudal, N. Sodium and health-concordance and controversy. BMJ 2020, 369, m2440. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.A.; Webster, J.; Dunford, E.K.; Jan, S.; Woodward, M.; Bolam, B.; Neal, B.; Trieu, K.; Reimers, J.; Armstrong, S.; et al. Sources of dietary sodium and implications for a statewide salt reduction initiative in Victoria, Australia. Br. J. Nutr. 2020, 123, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, L.; Galletti, F.; Strazzullo, P. Dietary salt intake and risk of gastric cancer. Cancer Treat. Res. 2014, 159, 83–95. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Cho, H.J.; Moon, S.; Choi, J.; Lee, S.; Ahn, C.; Yoo, K.Y.; Kim, I.; Ko, K.P.; Lee, J.E.; et al. Pickled Vegetable and Salted Fish Intake and the Risk of Gastric Cancer: Two Prospective Cohort Studies and a Meta-Analysis. Cancers 2020, 12, 996. [Google Scholar] [CrossRef]
- Wu, B.; Yang, D.; Yang, S.; Zhang, G. Dietary Salt Intake and Gastric Cancer Risk: A Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 801228. [Google Scholar] [CrossRef]
- Ning, F.L.; Lyu, J.; Pei, J.P.; Gu, W.J.; Zhang, N.N.; Cao, S.Y.; Zeng, Y.J.; Abe, M.; Nishiyama, K.; Zhang, C.D. The burden and trend of gastric cancer and possible risk factors in five Asian countries from 1990 to 2019. Sci. Rep. 2022, 12, 5980. [Google Scholar] [CrossRef] [PubMed]
- Caston, R.R.; Loh, J.T.; Voss, B.J.; McDonald, W.H.; Scholz, M.B.; McClain, M.S.; Cover, T.L. Effect of environmental salt concentration on the Helicobacter pylori exoproteome. J. Proteom. 2019, 202, 103374. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.C.; Dangler, C.A.; Taylor, N.S.; King, A.; Koh, T.J.; Wang, T.C. High-salt induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res. 1999, 59, 4823–4828. [Google Scholar] [PubMed]
- Furihata, C.; Ohta, H.; Katsuyama, T. Cause and effect between concentration-dependent tissue damage and temporary cell proliferation in rat stomach mucosa by NaCl, a stomach tumor promoter. Carcinogenesis 1996, 17, 401–406. [Google Scholar] [CrossRef]
- Xiao, F.; Crissey, M.A.; Lynch, J.P.; Kaestner, K.H.; Silberg, D.G.; Suh, E. Intestinal metaplasia with a high salt diet induces epithelial proliferation and alters cell composition in the gastric mucosa of mice. Cancer Biol. Ther. 2005, 4, 669–675. [Google Scholar] [CrossRef]
- Kountouras, J.; Zavos, C.; Chatzopoulos, D. Salt intake and Helicobacter pylori infection. J. Hypertens. 2004, 22, 2397. [Google Scholar] [CrossRef] [PubMed]
- De Koster, E.; Buset, M.; Fernandes, E.; Deltenre, M. Helicobacter pylori: The link with gastric cancer. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. (ECP) 1994, 3, 247–257. [Google Scholar] [CrossRef]
- Song, J.H.; Kim, Y.S.; Heo, N.J.; Lim, J.H.; Yang, S.Y.; Chung, G.E.; Kim, J.S. High Salt Intake Is Associated with Atrophic Gastritis with Intestinal Metaplasia. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1133–1138. [Google Scholar] [CrossRef]
- Tubbs, A.L.; Liu, B.; Rogers, T.D.; Sartor, R.B.; Miao, E.A. Dietary Salt Exacerbates Experimental Colitis. J. Immunol. 2017, 199, 1051–1059. [Google Scholar] [CrossRef]
- Vences-Mejía, A.; Caballero-Ortega, H.; Dorado-González, V.; Gamboa-Domínguez, A.; Gómez-Ruiz, C.; Camacho-Carranza, R.; Espinosa-Aguirre, J.J. Cytochrome P450 expression in rat gastric epithelium with intestinal metaplasia induced by high dietary NaCl levels. Environ. Toxicol. Pharmacol. 2005, 20, 57–64. [Google Scholar] [CrossRef]
- Maddineni, G.; Xie, J.J.; Brahmbhatt, B.; Mutha, P. Diet and carcinogenesis of gastric cancer. Curr. Opin. Gastroenterol. 2022, 38, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Tricker, A.R.; Pfundstein, B.; Theobald, E.; Preussmann, R.; Spiegelhalder, B. Mean daily intake of volatile N-nitrosamines from foods and beverages in West Germany in 1989–1990. Food Chem. Toxicol. 1991, 29, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Jakszyn, P.; Agudo, A.; Berenguer, A.; Ibáñez, R.; Amiano, P.; Pera, G.; Ardanaz, E.; Barricarte, A.; Chirlaque, M.D.; Dorronsoro, M.; et al. Intake and food sources of nitrites and N-nitrosodimethylamine in Spain. Public Health Nutr. 2006, 9, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Ohtake, K.; Uchida, H. NO-Rich Diet for Lifestyle-Related Diseases. Nutrients 2015, 7, 4911–4937. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tsukamoto, T.; Mizoshita, T.; Tanaka, H.; Kumagai, T.; Ota, H.; Katsuyama, T.; Asaka, M.; Tatematsu, M. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int. J. Cancer 2006, 119, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kim, K.; Lee, S.A.; Kwon, S.O.; Lee, J.K.; Keum, N.; Park, S.M. Effect of Red, Processed, and White Meat Consumption on the Risk of Gastric Cancer: An Overall and Dose-Response Meta-Analysis. Nutrients 2019, 11, 826. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Wu, L.; Guan, W. Dietary Nitrates, Nitrites, and Nitrosamines Intake and the Risk of Gastric Cancer: A Meta-Analysis. Nutrients 2015, 7, 9872–9895. [Google Scholar] [CrossRef]
- Zhang, F.X.; Miao, Y.; Ruan, J.G.; Meng, S.P.; Dong, J.D.; Yin, H.; Huang, Y.; Chen, F.R.; Wang, Z.C.; Lai, Y.F. Association Between Nitrite and Nitrate Intake and Risk of Gastric Cancer: A Systematic Review and Meta-Analysis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 1788–1799. [Google Scholar] [CrossRef]
- Compare, D.; Rocco, A.; Nardone, G. Risk factors in gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 302–308. [Google Scholar]
- Toh, J.W.T.; Wilson, R.B. Pathways of Gastric Carcinogenesis, Helicobacter pylori Virulence and Interactions with Antioxidant Systems, Vitamin C and Phytochemicals. Int. J. Mol. Sci. 2020, 21, 6451. [Google Scholar] [CrossRef]
- Kobayashi, J. Effect of diet and gut environment on the gastrointestinal formation of N-nitroso compounds: A review. Nitric Oxide Biol. Chem. 2018, 73, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.S.; Alexander, D.D.; Coughlin, J.R.; Milkowski, A.L.; Boffetta, P. Ingested nitrate and nitrite and stomach cancer risk: An updated review. Food Chem. Toxicol. 2012, 50, 3646–3665. [Google Scholar] [CrossRef] [PubMed]
- La Vecchia, C.; Negri, E.; Franceschi, S.; Decarli, A. Case-control study on influence of methionine, nitrite, and salt on gastric carcinogenesis in northern Italy. Nutr. Cancer 1997, 27, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Takahashi, M.; Fukushima, S.; Hananouchi, M.; Shirai, T. Effects in rats of sodium chloride on experimental gastric cancers induced by N-methyl-N-nitro-N-nitrosoguanidine or 4-nitroquinoline-1-oxide. J. Natl. Cancer Inst. 1975, 55, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Tsukamoto, T.; Hirano, N.; Mizoshita, T.; Kato, S.; Takasu, S.; Ban, H.; Tatematsu, M. Synergistic upregulation of inducible nitric oxide synthase and cyclooxygenase-2 in gastric mucosa of Mongolian gerbils by a high-salt diet and Helicobacter pylori infection. Histol. Histopathol. 2008, 23, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Berg, D.E. Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Investig. 2001, 107, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L. Helicobacter pylori Divers. Gastric Cancer Risk. mBio 2016, 7, e01869-15. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.T.; Beckett, A.C.; Scholz, M.B.; Cover, T.L. High-Salt Conditions Alter Transcription of Helicobacter pylori Genes Encoding Outer Membrane Proteins. Infect. Immun. 2018, 86, e00626-17. [Google Scholar] [CrossRef]
- Loh, J.T.; Gaddy, J.A.; Algood, H.M.; Gaudieri, S.; Mallal, S.; Cover, T.L. Helicobacter pylori adaptation in vivo in response to a high-salt diet. Infect. Immun. 2015, 83, 4871–4883. [Google Scholar] [CrossRef]
- Gancz, H.; Jones, K.R.; Merrell, D.S. Sodium chloride affects Helicobacter pylori growth and gene expression. J. Bacteriol. 2008, 190, 4100–4105. [Google Scholar] [CrossRef]
- Loh, J.T.; Torres, V.J.; Cover, T.L. Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res. 2007, 67, 4709–4715. [Google Scholar] [CrossRef] [PubMed]
- Beckett, A.C.; Loh, J.T.; Chopra, A.; Leary, S.; Lin, A.S.; McDonnell, W.J.; Dixon, B.R.E.A.; Noto, J.M.; Israel, D.A.; Peek, R.M., Jr.; et al. Helicobacter pylori genetic diversification in the Mongolian gerbil model. PeerJ 2018, 6, e4803. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.T.; Struttmann, E.L.; Favret, N.; Harvey, M.L.; Pakala, S.B.; Chopra, A.; McClain, M.S.; Cover, T.L. A Positively Selected fur-R88H Mutation Enhances Helicobacter pylori Fitness in a High-Salt Environment and Alters Fur-Dependent Regulation of Gene Expression. Infect. Immun. 2023, 91, e0042022. [Google Scholar] [CrossRef] [PubMed]
- Collatuzzo, G.; Pelucchi, C.; Negri, E.; López-Carrillo, L.; Tsugane, S.; Hidaka, A.; Shigueaki Hamada, G.; Hernández-Ramírez, R.U.; López-Cervantes, M.; Malekzadeh, R.; et al. Exploring the interactions between Helicobacter pylori (Hp) infection and other risk factors of gastric cancer: A pooled analysis in the Stomach cancer Pooling (StoP) Project. Int. J. Cancer 2021, 149, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; ALSalamat, H.A.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Alu, A.; Wei, Y.; Wei, X.; Luo, M. The modulatory effect of high salt on immune cells and related diseases. Cell Prolif. 2022, 55, e13250. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Matsui, H.; Nagano, Y.N.; Kaneko, T.; Indo, H.P.; Majima, H.J.; Hyodo, I. Salt is an oxidative stressor for gastric epithelial cells. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2013, 64, 89–94. [Google Scholar]
- Butcher, L.D.; den Hartog, G.; Ernst, P.B.; Crowe, S.E. Oxidative Stress Resulting From Helicobacter pylori Infection Contributes to Gastric Carcinogenesis. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 316–322. [Google Scholar] [CrossRef]
- Han, L.; Shu, X.; Wang, J. Helicobacter pylori-Mediated Oxidative Stress and Gastric Diseases: A Review. Front. Microbiol. 2022, 13, 811258. [Google Scholar] [CrossRef]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori Virulence Factors-Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.; Greenfield, L.K.; Bronte-Tinkew, D.; Capurro, M.I.; Rizzuti, D.; Jones, N.L. VacA promotes CagA accumulation in gastric epithelial cells during Helicobacter pylori infection. Sci. Rep. 2019, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Helicobacter pylori and gastric carcinogenesis. J. Gastroenterol. 2009, 44, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Ebik, B.; Aslan, N.; Ekin, N.; Bacaksiz, F.; Arpa, M.; Neselioglu, S.; Erel, O.; Ucmak, F. Oxidative stress and the importance of H. pylori eradication in patients with functional dyspepsia. Saudi J. Gastroenterol. 2022, 28, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.C.; Piazuelo, M.B.; Luis, P.B.; Barry, D.P.; Allaman, M.M.; Asim, M.; Sebrell, T.A.; Finley, J.L.; Rose, K.L.; Hill, S.; et al. Spermine oxidase mediates Helicobacter pylori-induced gastric inflammation, DNA damage, and carcinogenic signaling. Oncogene 2020, 39, 4465–4474. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, D.; McGinn, T.R.; Ye, X.; Bagchi, M.; Krohn, R.L.; Chatterjee, A.; Stohs, S.J. Helicobacter pylori-induced oxidative stress and DNA damage in a primary culture of human gastric mucosal cells. Dig. Dis. Sci. 2002, 47, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Thalmaier, U.; Lehn, N.; Pfeffer, K.; Stolte, M.; Vieth, M.; Schneider-Brachert, W. Role of tumor necrosis factor alpha in Helicobacter pylori gastritis in tumor necrosis factor receptor 1-deficient mice. Infect. Immun. 2002, 70, 3149–3155. [Google Scholar] [CrossRef]
- Lee, K.E.; Khoi, P.N.; Xia, Y.; Park, J.S.; Joo, Y.E.; Kim, K.K.; Choi, S.Y.; Jung, Y.D. Helicobacter pylori and interleukin-8 in gastric cancer. World J. Gastroenterol. 2013, 19, 8192–8202. [Google Scholar] [CrossRef]
- Lu, S.Y.; Guo, S.; Chai, S.B.; Yang, J.Q.; Yue, Y.; Li, H.; Sun, P.M.; Zhang, T.; Sun, H.W.; Zhou, J.L.; et al. Autophagy in Gastric Mucosa: The Dual Role and Potential Therapeutic Target. BioMed Res. Int. 2021, 2021, 2648065. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, W.; Yan, S.; Li, J.; Wei, H.; Zhao, W. CagA and VacA inhibit gastric mucosal epithelial cell autophagy and promote the progression of gastric precancerous lesions. Zhong Nan Da Xue Xue Bao. Yi Xue Ban = J. Cent. South Univ. Med. Sci. 2022, 47, 942–951. [Google Scholar] [CrossRef]
- Wüstner, S.; Anderl, F.; Wanisch, A.; Sachs, C.; Steiger, K.; Nerlich, A.; Vieth, M.; Mejías-Luque, R.; Gerhard, M. Helicobacter pylori γ-glutamyl transferase contributes to colonization and differential recruitment of T cells during persistence. Sci. Rep. 2017, 7, 13636. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed]
- Gaddy, J.A.; Radin, J.N.; Loh, J.T.; Zhang, F.; Washington, M.K.; Peek, R.M., Jr.; Algood, H.M.S.; Cover, T.L. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect. Immun. 2013, 81, 2258–2267. [Google Scholar] [CrossRef] [PubMed]
- Varga, M.G.; Wood, C.R.; Butt, J.; Ryan, M.E.; You, W.C.; Pan, K.; Waterboer, T.; Epplein, M.; Shaffer, C.L. Immunostimulatory membrane proteins potentiate H. pylori-induced carcinogenesis by enabling CagA translocation. Gut Microbes 2021, 13, 1862613. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Kanemitsu, A.; Knight, C.T.; Hatakeyama, M. Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell. Mol. Immunol. 2020, 17, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M., Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Yoshihashi, K.; Suzuki, H.; Tsutsumi, S.; Mutoh, H.; Maeda, S.; Yamagata, Y.; Seto, Y.; Aburatani, H.; Hatakeyama, M. CDX1 confers intestinal phenotype on gastric epithelial cells via induction of stemness-associated reprogramming factors SALL4 and KLF5. Proc. Natl. Acad. Sci. USA 2012, 109, 20584–20589. [Google Scholar] [CrossRef]
- Wei, J.; Noto, J.M.; Zaika, E.; Romero-Gallo, J.; Piazuelo, M.B.; Schneider, B.; El-Rifai, W.; Correa, P.; Peek, R.M.; Zaika, A.I. Bacterial CagA protein induces degradation of p53 protein in a p14ARF-dependent manner. Gut 2015, 64, 1040–1048. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.M.; Honjo, T.; Chiba, T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef]
- Zamperone, A.; Cohen, D.; Stein, M.; Viard, C.; Müsch, A. Inhibition of polarity-regulating kinase PAR1b contributes to Helicobacter pylori inflicted DNA Double Strand Breaks in gastric cells. Cell Cycle 2019, 18, 299–311. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Yao, Y.; Yan, W.; Park, D.I.; Kim, J.J.; Gooding, W.; Abudayyeh, S.; Graham, D.Y. CpG methylation and reduced expression of O6-methylguanine DNA methyltransferase is associated with Helicobacter pylori infection. Gastroenterology 2010, 138, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.; Chen, L.F. Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Yuan, Y. The Role of Gastric Mucosal Immunity in Gastric Diseases. J. Immunol. Res. 2020, 2020, 7927054. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Patel, G.K.; Ghoshal, U.C. Helicobacter pylori-Induced Inflammation: Possible Factors Modulating the Risk of Gastric Cancer. Pathogens 2021, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Díaz, P.; Valenzuela Valderrama, M.; Bravo, J.; Quest, A.F.G. Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front. Microbiol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gu, J.; Chen, J.; Zhang, P.; Ji, R.; Qian, H.; Xu, W.; Zhang, X. Interaction with neutrophils promotes gastric cancer cell migration and invasion by inducing epithelial-mesenchymal transition. Oncol. Rep. 2017, 38, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Dewayani, A.; Fauzia, K.A.; Alfaray, R.I.; Waskito, L.A.; Doohan, D.; Rezkitha, Y.A.A.; Abdurachman, A.; Kobayashi, T.; I’tishom, R.; Yamaoka, Y.; et al. The Roles of IL-17, IL-21, and IL-23 in the Helicobacter pylori Infection and Gastrointestinal Inflammation: A Review. Toxins 2021, 13, 315. [Google Scholar] [CrossRef]
- Lattanzi, G.; Strati, F.; Díaz-Basabe, A.; Perillo, F.; Amoroso, C.; Protti, G.; Rita Giuffrè, M.; Iachini, L.; Baeri, A.; Baldari, L.; et al. iNKT cell-neutrophil crosstalk promotes colorectal cancer pathogenesis. Mucosal Immunol. 2023, 16, 326–340. [Google Scholar] [CrossRef]
- Karkhah, A.; Ebrahimpour, S.; Rostamtabar, M.; Koppolu, V.; Darvish, S.; Vasigala, V.K.R.; Validi, M.; Nouri, H.R. Helicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases. Microbiol. Res. 2019, 218, 49–57. [Google Scholar] [CrossRef]
- Senchukova, M.A. Helicobacter pylori and Gastric Cancer Progression. Curr. Microbiol. 2022, 79, 383. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Camargo, M.C.; El-Omar, E.; Liou, J.M.; Peek, R.; Schulz, C.; Smith, S.I.; Suerbaum, S. Helicobacter pylori infection. Nat. Rev. Dis. Primers 2023, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Erah, P.O.; Goddard, A.F.; Barrett, D.A.; Shaw, P.N.; Spiller, R.C. The stability of amoxycillin, clarithromycin and metronidazole in gastric juice: Relevance to the treatment of Helicobacter pylori infection. J. Antimicrob. Chemother. 1997, 39, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Villoria, A.; Garcia, P.; Calvet, X.; Gisbert, J.P.; Vergara, M. Meta-analysis: High-dose proton pump inhibitors vs. standard dose in triple therapy for Helicobacter pylori eradication. Aliment. Pharmacol. Ther. 2008, 28, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Fallone, C.A.; Chiba, N.; van Zanten, S.V.; Fischbach, L.; Gisbert, J.P.; Hunt, R.H.; Jones, N.L.; Render, C.; Leontiadis, G.I.; Moayyedi, P.; et al. The Toronto Consensus for the Treatment of Helicobacter pylori Infection in Adults. Gastroenterology 2016, 151, 51–69.e14. [Google Scholar] [CrossRef] [PubMed]
- Nyssen, O.P.; Bordin, D.; Tepes, B.; Pérez-Aisa, Á.; Vaira, D.; Caldas, M.; Bujanda, L.; Castro-Fernandez, M.; Lerang, F.; Leja, M.; et al.; et al. Hp-EuReg Investigators European Registry on Helicobacter pylori management (Hp-EuReg): Patterns and trends in first-line empirical eradication prescription and outcomes of 5 years and 21,533 patients. Gut 2021, 70, 40–54. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Bazzoli, F.; Delchier, J.C.; Celiñski, K.; Giguère, M.; Rivière, M.; Mégraud, F. Pylera Study Group Helicobacter pylori eradication with a capsule containing bismuth subcitrate potassium, metronidazole, and tetracycline given with omeprazole versus clarithromycin-based triple therapy: A randomised, open-label, non-inferiority, phase 3 trial. Lancet 2011, 377, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.T.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. Management of Helicobacter pylori infection-the Maastricht V/Florence Consensus Report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef]
- Saleem, N.; Howden, C.W. Update on the Management of Helicobacter pylori Infection. Curr. Treat. Options Gastroenterol. 2020, 18, 476–487. [Google Scholar] [CrossRef]
- Romano, M.; Cuomo, A.; Gravina, A.G.; Miranda, A.; Iovene, M.R.; Tiso, A.; Sica, M.; Rocco, A.; Salerno, R.; Marmo, R.; et al. Empirical levofloxacin-containing versus clarithromycin-containing sequential therapy for Helicobacter pylori eradication: A randomised trial. Gut 2010, 59, 1465–1470. [Google Scholar] [CrossRef]
- Megraud, F.; Bruyndonckx, R.; Coenen, S.; Wittkop, L.; Huang, T.D.; Hoebeke, M.; Bénéjat, L.; Lehours, P.; Goossens, H.; Glupczynski, Y. European Helicobacter pylori Antimicrobial Susceptibility Testing Working Group Helicobacter pylori resistance to antibiotics in Europe in 2018 and its relationship to antibiotic consumption in the community. Gut 2021, 70, 1815–1822. [Google Scholar] [CrossRef]
- Liou, J.M.; Fang, Y.J.; Chen, C.C.; Bair, M.J.; Chang, C.Y.; Lee, Y.C.; Chen, M.J.; Chen, C.C.; Tseng, C.H.; Hsu, Y.C.; et al. Taiwan Gastrointestinal Disease and Helicobacter Consortium Concomitant, bismuth quadruple, and 14-day triple therapy in the first-line treatment of Helicobacter pylori: A multicentre, open-label, randomised trial. Lancet 2016, 388, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Guillemard, E.; Poirel, M.; Schäfer, F.; Quinquis, L.; Rossoni, C.; Keicher, C.; Wagner, F.; Szajewska, H.; Barbut, F.; Derrien, M.; et al. A Randomised, Controlled Trial: Effect of a Multi-Strain Fermented Milk on the Gut Microbiota Recovery after Helicobacter pylori Therapy. Nutrients 2021, 13, 3171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Cao, X.Y.; Zhu, H.L.; Miao, L. Comparative effectiveness of different probiotics supplements for triple Helicobacter pylori eradication: A network meta-analysis. Front. Cell. Infect. Microbiol. 2023, 13, 1120789. [Google Scholar] [CrossRef] [PubMed]
- Homan, M.; Orel, R. Are probiotics useful in Helicobacter pylori eradication? World J. Gastroenterol. 2015, 21, 10644–10653. [Google Scholar] [CrossRef] [PubMed]
- Francavilla, R.; Polimeno, L.; Demichina, A.; Maurogiovanni, G.; Principi, B.; Scaccianoce, G.; Ierardi, E.; Russo, F.; Riezzo, G.; Di Leo, A.; et al. Lactobacillus reuteri strain combination in Helicobacter pylori infection: A randomized, double-blind, placebo-controlled study. J. Clin. Gastroenterol. 2014, 48, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Srisuphanunt, M.; Wilairatana, P.; Kooltheat, N.; Duangchan, T.; Katzenmeier, G.; Rose, J.B. Molecular Mechanisms of Antibiotic Resistance and Novel Treatment Strategies for Helicobacter pylori Infections. Trop. Med. Infect. Dis. 2023, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, I.; Chua, E.G.; Tay, A.C.Y.; Kostrešević, A.; Pavlović, B.; Jončić Savić, K. Savory, Oregano and Thyme Essential Oil Mixture (HerbELICO®) Counteracts Helicobacter pylori. Molecules 2023, 28, 2138. [Google Scholar] [CrossRef]
- Salehi, B.; Sharopov, F.; Martorell, M.; Rajkovic, J.; Ademiluyi, A.O.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Iriti, M.; Sharifi-Rad, J. Phytochemicals in Helicobacter pylori Infections: What Are We Doing Now? Int. J. Mol. Sci. 2018, 19, 2361. [Google Scholar] [CrossRef]
- Wu, X.; Chen, L.; Cheng, J.; Qian, J.; Fang, Z.; Wu, J. Effect of Dietary Salt Intake on Risk of Gastric Cancer: A Systematic Review and Meta-Analysis of Case-Control Studies. Nutrients 2022, 14, 4260. [Google Scholar] [CrossRef]
- Ge, S.; Feng, X.; Shen, L.; Wei, Z.; Zhu, Q.; Sun, J. Association between Habitual Dietary Salt Intake and Risk of Gastric Cancer: A Systematic Review of Observational Studies. Gastroenterol. Res. Pr. 2012, 2012, 808120. [Google Scholar] [CrossRef]
- Peleteiro, B.; Lopes, C.; Figueiredo, C.; Lunet, N. Salt intake and gastric cancer risk according to Helicobacter pylori infection, smoking, tumour site and histological type. Br. J. Cancer 2011, 104, 198–207. [Google Scholar] [CrossRef]
- Zhong, C.; Li, K.N.; Bi, J.W.; Wang, B.C. Sodium intake, salt taste and gastric cancer risk according to helicobacter pylori infection, smoking, histological type and tumor sit in China. Asian Pac. J. Cancer Prev. 2012, 13, 2481–2484. [Google Scholar] [CrossRef][Green Version]


| Therapy Name | Dosing | Duration (Days) | Eradication (%) |
|---|---|---|---|
| Clarithromycin-based PPI-TT | PPI bid + clarithromycin 500 mg bid + amoxicillin 1 gr bid or metronidazole 500 mg tad | 14 | 70–85 |
| Bismuth-based quadruple | PPI bid + bismuth salicylate 300 mg qid + metronidazole 500 mg tid + tetracycline 500 mg qid | 14 | 75–90 |
| PPI-TTT | PPI bid + amoxicillin 1 gr bid + clarithromycin 500 mg bid + metronidazole 500 mg tid | 14 | 90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balendra, V.; Amoroso, C.; Galassi, B.; Esposto, J.; Bareggi, C.; Luu, J.; Scaramella, L.; Ghidini, M. High-Salt Diet Exacerbates H. pylori Infection and Increases Gastric Cancer Risks. J. Pers. Med. 2023, 13, 1325. https://doi.org/10.3390/jpm13091325
Balendra V, Amoroso C, Galassi B, Esposto J, Bareggi C, Luu J, Scaramella L, Ghidini M. High-Salt Diet Exacerbates H. pylori Infection and Increases Gastric Cancer Risks. Journal of Personalized Medicine. 2023; 13(9):1325. https://doi.org/10.3390/jpm13091325
Chicago/Turabian StyleBalendra, Vyshnavy, Chiara Amoroso, Barbara Galassi, Josephine Esposto, Claudia Bareggi, Jennie Luu, Lucia Scaramella, and Michele Ghidini. 2023. "High-Salt Diet Exacerbates H. pylori Infection and Increases Gastric Cancer Risks" Journal of Personalized Medicine 13, no. 9: 1325. https://doi.org/10.3390/jpm13091325
APA StyleBalendra, V., Amoroso, C., Galassi, B., Esposto, J., Bareggi, C., Luu, J., Scaramella, L., & Ghidini, M. (2023). High-Salt Diet Exacerbates H. pylori Infection and Increases Gastric Cancer Risks. Journal of Personalized Medicine, 13(9), 1325. https://doi.org/10.3390/jpm13091325