
Modulation of Radiation Doses and Chimeric Antigen Receptor T Cells: A Promising New Weapon in Solid Tumors—A Narrative Review

 , , , ,
, , , ,
Abstract
1. Introduction
2. CART: Chimeric Antigen Receptor (CAR) T Cells
2.1. CART Cell Structure
2.2. Toxicities
2.2.1. On-Target On-Tumor Toxicity: Cytokine Release Syndrome
2.2.2. On-Target Off-Tumor Toxicity
2.2.3. Off-Target Toxicity
2.2.4. Neurotoxicity: ICANS (Immune-Effector-Cell-Associated Neurotoxicity Syndrome)
2.2.5. Immunogenicity: Anaphylaxis
3. Tumor Microenvironment (TME)
4. Radiotherapy
4.1. LATTICE Therapy
4.1.1. LATTICE Technique
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Linnemann, C.; Mezzadra, R.; Schumacher, T.N.M. TCR repertoires of intratumoral T-cell subsets. Immunol. Rev. 2013, 257, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Blattman, J.N.; Greenberg, P.D. Cancer Immunotherapy: A Treatment for the Masses. Science 2004, 305, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. Does the Immune System See Tumors as Foreign or Self? Annu. Rev. Immunol. 2003, 21, 807–839. [Google Scholar] [CrossRef]
- Traversari, C.; van der Bruggen, P.; Luescher, I.F.; Lurquin, C.; Chomez, P.; Van Pel, A.; De Plaen, E.; Amar-Costesec, A.; Boon, T. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J. Exp. Med. 1992, 176, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Stone, B.; Schummer, M.; Paley, P.J.; Crawford, M.; Ford, M.; Urban, N.; Nelson, B.H. MAGE-F1, a novel ubiquitously expressed member of the MAGE superfamily. Gene 2001, 267, 173–182. [Google Scholar] [CrossRef]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Model. Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: Review of the literature and challenges ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy with TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lym-phoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress after Allogeneic Hematopoietic Stem-Cell Transplantation without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Johnson, L.A.; Heemskerk, B.; Powell, D.J.; Cohen, C.J.; Morgan, R.A.; Dudley, M.E.; Robbins, P.F.; Rosenberg, S.A. Gene Transfer of Tumor-Reactive TCR Confers Both High Avidity and Tumor Reactivity to Nonreactive Peripheral Blood Mononuclear Cells and Tumor-Infiltrating Lymphocytes. J. Immunol. 2006, 177, 6548–6559. [Google Scholar] [CrossRef]
- Cartellieri, M.; Bachmann, M.; Feldmann, A.; Bippes, C.; Stamova, S.; Wehner, R.; Temme, A.; Schmitz, M. Chimeric Antigen Receptor-Engineered T Cells for Immunotherapy of Cancer. J. Biomed. Biotechnol. 2010, 2010, 956304. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Brandjes, B.D.; Davila, M.L. Adding chimeric antigen receptor induced killer cells to the medical oncology shelf. J. Clin. Investig. 2019, 129, 5077–5078. [Google Scholar] [CrossRef] [PubMed]
- Brocker, T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 2000, 96, 1999–2001. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Li, H.; Tang, B. Radiotherapy plus CAR-T cell therapy to date: A note for cautions optimism? Front. Immunol. 2022, 13, 1033512. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.C.; Latouche, J.B.; Krause, A.; Heston, W.D.; Bander, N.H.; Sadelain, M. Cancer patient T cells genetically targeted to prostate specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific mem-brane antigen. Neoplasia 1999, 1, 123–127. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Fujiwara, K.; Kitaura, M.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Okada, N. Structure of the signal transduction domain in sec-ond generation CAR regulates the input efficiency of CAR signals. Int. J. Mol. Sci. 2021, 22, 2476. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T cell therapy for cancer in the clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Jain, T.; Santomasso, B.D.; Mead, E.; Wudhikarn, K.; Silverberg, M.L.; Batlevi, Y.; Shouval, R.; Devlin, S.M.; Batlevi, C.; et al. Comparing CAR T-cell toxicity grading systems: Application of the ASTCT grading system and implications for management. Blood Adv. 2020, 4, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2017, 15, 47–62. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Bugelski, P.J.; Achuthanandam, R.; Capocasale, R.J.; Treacy, G.; Bouman-Thio, E. Monoclonal antibodyinduced cyto-kine-release syndrome. Expert Rev. Clin. Immunol. 2009, 5, 499–521. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Weiss, S.L.M.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome after Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to ab-normal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing Cytokine Release Syndrome Associated with Novel T Cell-Engaging Therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef]
- De Benedetti, H.; Brunner, I.; Ruperto, N.; Kenwright, A.; Wright, S.; Calvo, I.; Cuttica, R.; Ravelli, A.; Schneider, R.; Woo, P.; et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2385–2395. [Google Scholar] [CrossRef]
- Fisher, J.; Abramowski, P.; Don, N.D.W.; Flutter, B.; Capsomidis, A.; Cheung, G.W.-K.; Gustafsson, K.; Anderson, J. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol. Ther. 2017, 25, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-Derived HLA-A1–Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3–Directed T Cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Mei, H.; Jiang, H.; Wu, Y.; Guo, T.; Xia, L.; Jin, R.; Hu, Y. Neurological toxicities and coagulation disorders in the cytokine release syndrome during CAR-T therapy. Br. J. Haematol. 2017, 181, 689–692. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.K.; Turtle, C.J. Assessment and management of cytokine release syndrome and neurotoxicity following CD19 CAR-T cell therapy. Expert Opin. Biol. Ther. 2020, 20, 653–664. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint blockade reverses anergy in IL-13Rα2 Humanized scFv-Based CAR T cells to treat murine and canine gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Antoñana-Vildosola, A.; Zanetti, S.R.; Palazon, A. Enabling CAR-T cells for solid tumors: Rage against the suppressive tumor microenvironment. Int. Rev. Cell Mol. Biol. 2022, 37, 123–147. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordao, M.J.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Babar, Q.; Saeed, A.; Tabish, T.A.; Sarwar, M.; Thorat, N.D. Targeting the tumor microenvironment: Potential strategy for cancer therapeutics. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2023, 1869, 166746. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Bożyk, A.; Wojas-Krawczyk, K.; Krawczyk, P.; Milanowski, J. Tumor Microenvironment—A Short Review of Cellular and Interaction Diversity. Biology 2022, 11, 929. [Google Scholar] [CrossRef]
- Rossi, M.; Altomare, E.; Botta, C.; Cantafio, M.E.G.; Sarvide, S.; Caracciolo, D.; Riillo, C.; Gaspari, M.; Taverna, D.; Conforti, F.; et al. miR-21 antagonism abrogates Th17 tumor promoting functions in multiple myeloma. Leukemia 2020, 35, 823–834. [Google Scholar] [CrossRef]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef]
- Lee, J.; Lozano-Ruiz, B.; Yang, F.M.; Fan, D.D.; Shen, L.; González-Navajas, J.M. The Multifaceted Role of Th1, Th9, and Th17 Cells in Immune Checkpoint Inhibition Therapy. Front. Immunol. 2021, 12, 625667. [Google Scholar] [CrossRef]
- Jeske, S.S.; Weissinger, S.E.; Veit, J.A.; Brunner, C.; Huber, U.; Theodoraki, M.N.; Hoffmann, T.K.; Schuler, P.J.; Doescher, J. Treatment-induced changes of lymphocyte subsets in patients with adenoid cystic carcinoma of the head and neck. Eur. Arch. Oto-Rhino-Laryngol. 2019, 276, 1465–1473. [Google Scholar] [CrossRef]
- De Guillebon, E.; Dardenne, A.; Saldmann, A.; Séguier, S.; Tran, T.; Paolini, L.; Lebbe, C.; Tartour, E. Beyond the concept of cold and hot tumors for the development of novel predictive biomarkers and the rational design of immunotherapy combination. Int. J. Cancer 2020, 147, 1509–1518. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, L.L.; Gorris, M.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Forster, J.C.; Marcu, L.G.; Bezak, E. Approaches to combat hypoxia in cancer therapy and the potential for in silico models in their evaluation. Phys. Medica 2019, 64, 145–156. [Google Scholar] [CrossRef]
- Xie, G.; Liu, Y.; Yao, Q.; Zheng, R.; Zhang, L.; Lin, J.; Guo, Z.; Du, S.; Ren, C.; Yuan, Q.; et al. Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells. Sci. Rep. 2017, 7, 42396. [Google Scholar] [CrossRef]
- Farc, O.; Cristea, V. An overview of the tumor microenvironment, from cells to complex networks (Review). Exp. Ther. Med. 2020, 21, 96. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef]
- Iacobini, C.; Vitale, M.; Pugliese, G.; Menini, S. The “sweet” path to cancer: Focus on cellular glucose metabolism. Front. Oncol. 2023, 13, 1202093. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. Angiogenesis as a hallmark of solid tumors clinical perspectives. Cell Oncol. 2021, 44, 715–737. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liao, Q.; Zhao, C.; Zhu, C.; Feng, M.; Liu, Z.; Jiang, L.; Zhang, L.; Ding, X.; Yuan, M.; et al. Conditioned CAR T cells by hypoxia inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains anti-tumor efficacy. J. Immunother. Cancer 2021, 9, e002755. [Google Scholar] [CrossRef]
- Overgaard, J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck—A systematic review and meta-analysis. Radiother. Oncol. 2011, 100, 22–32. [Google Scholar] [CrossRef]
- Rodriguez Garcia, A.; Palazon, A.; Noguera Ortega, E.; Powell, D.J.; Guedan, S. CAR T cells Hit the tumor mi-croenvironment: Strategies to overcome tumor escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells In Vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, srep39833. [Google Scholar] [CrossRef]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Gladney, W.; Plesa, G.; Vapiwala, N.; Carpenter, E.; Maude, S.L.; Lal, P.; Lacey, S.F.; Melenhorst, J.J.; Sebro, R.; et al. A phase I clinical trial of PSMA-directed/TGFβ-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2019, 37, TPS347. [Google Scholar] [CrossRef]
- Dorff, T.B.; Blanchard, S.; Carruth, P.; Wagner, J.; Kuhn, P.; Chaudhry, A.; Adkins, L.; Thomas, S.; Martirosyan, H.; Chu, P.; et al. A phase I study to evaluate PSCA-targeting chimeric antigen receptor (CAR)-T cells for patients with PSCA+ metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, TPS250. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rusch, V.W.; O’Cearbhaill, R.; Zhu, A.; Ngai, D.; McGee, E.; Chintala, N.; Messinger, J.; Cheema, W.; et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. J. Clin. Oncol. 2019, 37, 2511. [Google Scholar] [CrossRef]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef]
- Pai, S.I.; Cesano, A. Tumor Microenvironment. In Physiology & Behavior; Lee, P.P., Marincola, F.M., Eds.; Springer International Publishing: Cham, Switzerland, 2020; Volume 180, pp. 139–148. [Google Scholar]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef]
- Seyedin, S.N.; Schoenhals, J.E.; Lee, D.A.; Cortez, M.A.; Wang, X.; Niknam, S.; Tang, C.; Hong, D.S.; Naing, A.; Sharma, P.; et al. Strategies for combining immunotherapy with radiation for anticancer therapy. Immunotherapy 2015, 7, 967–980. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, Q.; Zheng, Z.; Liu, S.; Meng, L.; Dong, L.; Jiang, X. Vascular normalization in immunotherapy: A promising mecha nisms combined with radiotherapy. Biomed. Pharmacother. 2021, 139, 111607. [Google Scholar] [CrossRef] [PubMed]
- Hauth, F.; Ho, A.Y.; Ferrone, S.; Duda, D.G. Radiotherapy to enhance chimeric antigen receptor T cell therapeutic efficacy in solid tumors: A narrative review. JAMA Oncol. 2021, 7, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Chruściel, E.; Urban-Wójciuk, Z.; Arcimowicz, Ł.; Kurkowiak, M.; Kowalski, J.; Gliwiński, M.; Marjański, T.; Rzyman, W.; Biernat, W.; Dziadziuszko, R.; et al. Adoptive cell therapy—Harnessing antigen-specific t cells to target solid tumours. Cancers 2020, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef]
- Walle, T.; Monge, R.M.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther. Adv. Med. Oncol. 2018, 10, 1–27. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming Growth Factor-β Controls Development, Homeostasis, and Tolerance of T Cells by Regulatory T Cell-Dependent and -Independent Mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of Local Radiation Therapy in Cancer Immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Ruiz, M.E.; Garasa, S.; Rodriguez, I.; Solorzano, J.L.; Barbes, B.; Yanguas, A.; Teijeira, A.; Etxeberria, I.; Aristu, J.J.; Halin, C.; et al. Intercellular adhesion Molecule 1 and vascular cell adhesion molecule are induced by ionizing radiation on lymphatic endo-thelium. Int. J. Radiat. Oncol. Biol. Phys. 2017, 97, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Karin, N. The multiple faces of CXCL12 (SDF 1alpha) in the regulation of immunity during health and disease. J. Leukoc. Biol. 2010, 88, 463–473. [Google Scholar] [CrossRef]
- Li, C.G.; He, M.R.; Wu, F.L.; Li, Y.J.; Sun, A.M. Akt promotes irradiation induced regulatory T cell survival in hepatocellular car-cinoma. Am. J. Med. Sci. 2013, 346, 123–127. [Google Scholar] [CrossRef]
- Park, H.-R.; Jung, U. Depletion of NK Cells Resistant to Ionizing Radiation Increases Mutations in Mice after Whole-body Irradiation. In Vivo 2021, 35, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Pontoriero, A.; Critelli, P.; Conti, A.; Cardali, S.; Angileri, F.F.; Germanò, A.; Lillo, S.; Carretta, A.; Brogna, A.; Santacaterina, A.; et al. The “Combo” radiotherapy treatment for high-risk grade 2 meningiomas: Dose escalation and initial safety and efficacy analysis. J. Neuro-Oncol. 2022, 161, 203–214. [Google Scholar] [CrossRef]
- Parisi, S.; Ferini, G.; Cacciola, A.; Lillo, S.; Tamburella, C.; Santacaterina, A.; Bottari, A.; Brogna, A.; Ferrantelli, G.; Pontoriero, A.; et al. A non-surgical COMBO-therapy approach for locally advanced unresectable pancreatic adenocarcinoma: Preliminary results of a prospective study. Radiol. Medica 2022, 127, 214–219. [Google Scholar] [CrossRef]
- DeSelm, C.; Palomba, M.L.; Yahalom, J.; Hamieh, M.; Eyquem, J.; Rajasekhar, V.K.; Sadelain, M. Low dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol. Ther. 2018, 26, 2542–2552. [Google Scholar] [CrossRef]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, J.D.; Giobbie-Hurder, A.; Ranasinghe, S.; Kao, K.Z.; Lako, A.; Tsuji, J.; Liu, Y.; Brennick, R.C.; Gentzler, R.D.; Lee, C.; et al. Durvalumab plus tremelimumab alone or in combination with low-dose or hypofractionated radiotherapy in metastatic non-small-cell lung cancer refractory to previous PD(L)-1 therapy: An open-label, multicentre, randomised, phase 2 trial. Lancet Oncol. 2022, 23, 279–291. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Giobbie-Hurder, A.; Lako, A.; Thrash, E.M.; Brennick, R.C.; Kao, K.Z.; Manuszak, C.; Gentzler, R.D.; Tesfaye, A.; Jabbour, S.K.; et al. A Randomized Trial of Combined PD-L1 and CTLA-4 Inhibition with Tar-geted Low-Dose or Hypofractionated Radiation for Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2470–2480. [Google Scholar] [CrossRef]
- Meng, Y.; Beckett, M.A.; Liang, H.; Mauceri, H.J.; van Rooijen, N.; Cohen, K.S.; Weichselbaum, R.R. Blockade of Tumor Necrosis Factor α Signaling in Tumor-Associated Macrophages as a Radiosensitizing Strategy. Cancer Res. 2010, 70, 1534–1543. [Google Scholar] [CrossRef]
- Zammarchi, F.; Havenith, K.; Bertelli, F.; Vijayakrishnan, B.; Chivers, S.; van Berkel, P.H. CD25 targeted antibody drug conjugate depletes regulatory T cells and eliminates established syngeneic tumors via antitumor immunity. J. Immunother. Cancer 2020, 8, e000860. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.; Hagekyriakou, J.; Chindris, I.; Albarakati, H.; Leong, T.; Schlenker, R.; Keam, S.P.; Williams, S.G.; Neeson, P.J.; Johnstone, R.W.; et al. Regulatory T cells shape the differential impact of radiation dose fractionation schedules on host innate and adaptive antitumor immune defenses. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Laurent, P.A.; Morel, D.; Meziani, L.; Depil, S.; Deutsch, E. Radiotherapy as a means to increase the efficacy of T cell therapy in solid tumors. Oncoimmunology 2023, 12, 2158013. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, A.; Ahmed, M.M.; Mohiuddin, M.; Coleman, C.N. Exploiting sensitization windows of opportunity in hyper and hypo-fractionated radiation therapy. J. Thorac. Dis. 2014, 6, 287–302. [Google Scholar] [CrossRef]
- Morisada, M.; Clavijo, P.E.; Moore, E.; Sun, L.; Chamberlin, M.; Van Waes, C.; Hodge, J.W.; Mitchell, J.B.; Friedman, J.; Allen, C.T. PD-1 blockade reverses adaptive immune resistance induced by high-dose hypofractionated but not low-dose daily fractionated radiation. OncoImmunology 2018, 7, e1395996. [Google Scholar] [CrossRef]
- Navarro-Martín, A.; Galiana, I.L.; Frances, M.A.B.; Cacicedo, J.; Cortés, R.C.; Anton, S.C.; Sánchez, S.P.; Cuevas, S.B.; Parry, R.; Edo, F.G. Preliminary Study of the Effect of Stereotactic Body Radiotherapy (SBRT) on the Immune System in Lung Cancer Patients Unfit for Surgery: Immunophenotyping Analysis. Int. J. Mol. Sci. 2018, 19, 3963. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Barsoumian, H.B.; Ramapriyan, R.; Younes, A.I.; Caetano, M.S.; Menon, H.; Comeaux, N.I.; Cushman, T.R.; Schoenhals, J.E.; Cadena, A.P.; Reilly, T.P.; et al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J. Immunother. Cancer 2020, 8, e000537. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; Barsoumian, H.; Verma, V.; Cortez, M.A.; Welsh, J.W. Low-Dose Radiation Decreases Cancer-Associated Fibro-blasts and May Increase T-Cell Trafficking into Tumors. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, e530–e531. [Google Scholar] [CrossRef]
- Kim, R.-K.; Kim, M.-J.; Seong, K.M.; Kaushik, N.; Suh, Y.; Yoo, K.-C.; Cui, Y.-H.; Jin, Y.W.; Nam, S.Y.; Lee, S.-J. Beneficial effects of low dose radiation in response to the oncogenic KRAS induced cellular transformation. Sci. Rep. 2015, 5, 15809. [Google Scholar] [CrossRef]
- Donlon, N.E.; Power, R.; Hayes, C.; Reynolds, J.V.; Lysaght, J. Radiotherapy, immunotherapy, and the tumour microenvi-ron ment: Turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021, 502, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, G.; Reinwald, S.; Edwards, J.; Wong, N.; Yu, D.; Ward, R.; Smith, R.; Haydon, A.; Au, P.M.; van Zelm, M.C.; et al. Blood T cell profiling in metastatic melanoma patients as a marker for response to immune checkpoint inhibitors combined with radio therapy. Radiother. Oncol. 2022, 173, 299–305. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer im-mu notherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Rotte, A.; Frigault, M.J.; Ansari, A.; Gliner, B.; Heery, C.; Shah, B. Dose–response correlation for CAR-T cells: A systematic review of clinical studies. J. Immunother. Cancer 2022, 10, e005678. [Google Scholar] [CrossRef]
- Ferini, G.; Parisi, S.; Lillo, S.; Viola, A.; Minutoli, F.; Critelli, P.; Valenti, V.; Illari, S.I.; Brogna, A.; Umana, G.E.; et al. Impressive Results after “Metabolism-Guided” Lattice Irradiation in Patients Submitted to Palliative Radiation Therapy: Preliminary Results of LATTICE_01 Multicenter Study. Cancers 2022, 14, 3909. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, X.; Zhang, J.; Li, W.; Dong, F.; Chen, C.; Lin, Q.; Zhang, C.; Zheng, F.; Yan, W.; et al. Combined High-Dose LATTICE Radiation Therapy and Immune Checkpoint Blockade for Advanced Bulky Tumors: The Concept and a Case Report. Front. Oncol. 2021, 10, 548132. [Google Scholar] [CrossRef]
- Ferini, G.; Valenti, V.; Tripoli, A.; Illari, S.I.; Molino, L.; Parisi, S.; Cacciola, A.; Lillo, S.; Giuffrida, D.; Pergolizzi, S. Lattice or Oxygen-Guided Radiotherapy: What If They Converge? Possible Future Directions in the Era of Immunotherapy. Cancers 2021, 13, 3290. [Google Scholar] [CrossRef]
- Duriseti, S.; Kavanaugh, J.; Goddu, S.; Price, A.; Knutson, N.; Reynoso, F.; Michalski, J.; Mutic, S.; Robinson, C.; Spraker, M.B. Spatially fractionated stereotactic body radiation therapy (Lattice) for large tumors. Adv. Radiat. Oncol. 2021, 6, 100639. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Perez, N.C.; Zheng, Y.; Li, X.; Jiang, L.; Amendola, B.E.; Xu, B.; Mayr, N.A.; Lu, J.J.; Hatoum, G.F.; et al. The Technical and Clinical Implementation of LATTICE Radiation Therapy (LRT). Radiat. Res. 2020, 194, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Blanco Suarez, J.M.; Amendola, B.E.; Perez, N.; Amendola, M.; Wu, X. The use of Lattice radiation therapy (LRT) in the treatment of bulky tumors: A case report of a large metastatic mixed mullerian ovarian tumor. Cureus 2015, 7, e389. [Google Scholar] [CrossRef] [PubMed]
- Ertan, F.; Yeginer, M.; Zorlu, F. Dosimetric Performance Evaluation of MLC-based and Cone-based 3D Spatially Fractionated LATTICE Radiotherapy. Radiat. Res. 2023, 199, 161–169. [Google Scholar] [CrossRef]


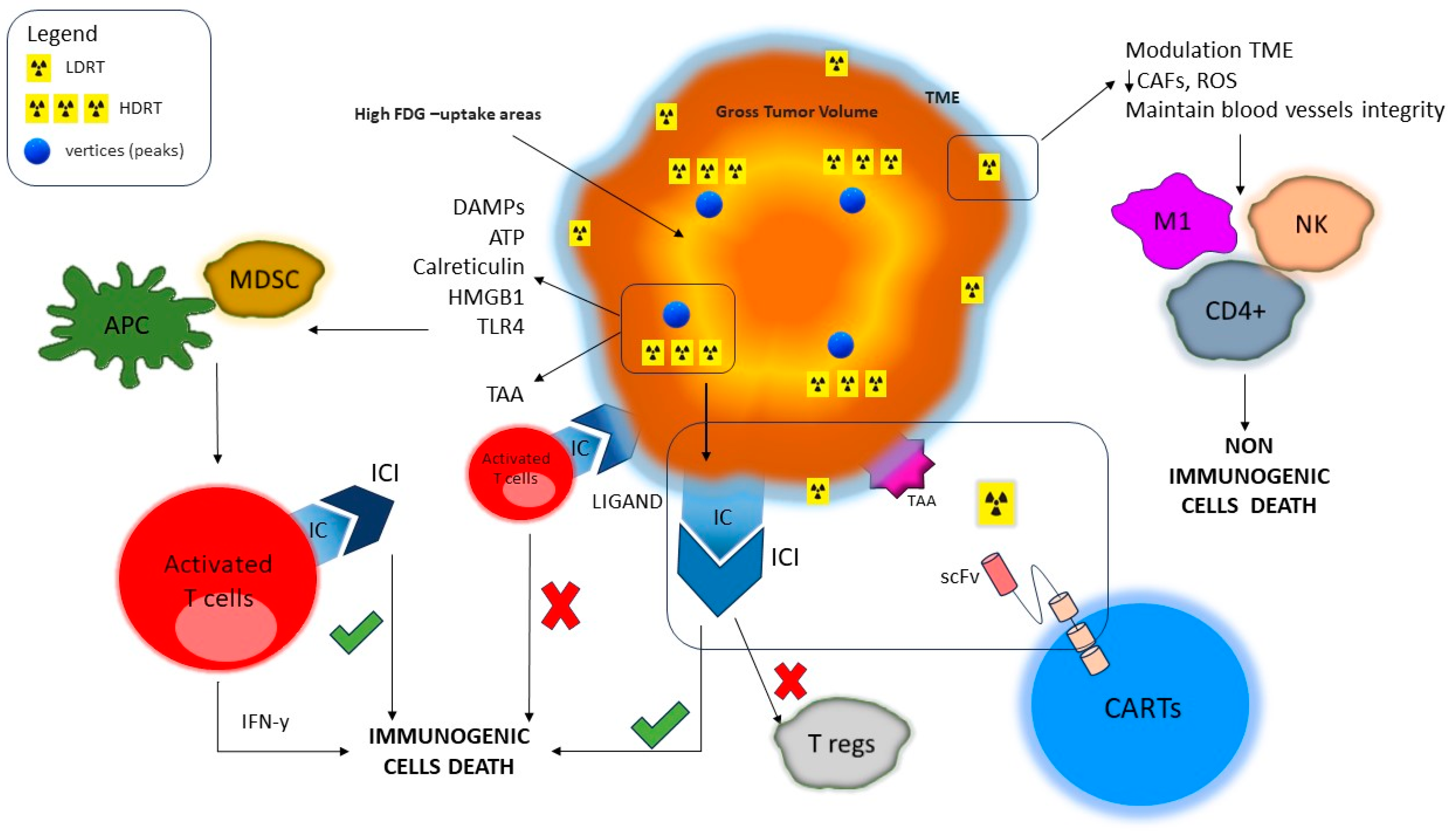{kind=link}
| T Cell Infiltration | Mutation Burden and PD-L1 Expression | Response to Immunotherapy | Example | |
|---|---|---|---|---|
| Hot Tumor |
| High mutation burden and high expression of PD-L1 | Sensitive | Melanoma, non-small-cell lung cancer, and cancers of the bladder, head and neck, kidney, liver |
| Cold Tumor |
| Low mutation burden and low expression of PD-L1 | Resistant | Breast, ovary, prostate, pancreas, brain glioblastoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontoriero, A.; Critelli, P.; Chillari, F.; Ferrantelli, G.; Sciacca, M.; Brogna, A.; Parisi, S.; Pergolizzi, S. Modulation of Radiation Doses and Chimeric Antigen Receptor T Cells: A Promising New Weapon in Solid Tumors—A Narrative Review. J. Pers. Med. 2023, 13, 1261. https://doi.org/10.3390/jpm13081261
Pontoriero A, Critelli P, Chillari F, Ferrantelli G, Sciacca M, Brogna A, Parisi S, Pergolizzi S. Modulation of Radiation Doses and Chimeric Antigen Receptor T Cells: A Promising New Weapon in Solid Tumors—A Narrative Review. Journal of Personalized Medicine. 2023; 13(8):1261. https://doi.org/10.3390/jpm13081261
Chicago/Turabian StylePontoriero, Antonio, Paola Critelli, Federico Chillari, Giacomo Ferrantelli, Miriam Sciacca, Anna Brogna, Silvana Parisi, and Stefano Pergolizzi. 2023. "Modulation of Radiation Doses and Chimeric Antigen Receptor T Cells: A Promising New Weapon in Solid Tumors—A Narrative Review" Journal of Personalized Medicine 13, no. 8: 1261. https://doi.org/10.3390/jpm13081261
APA StylePontoriero, A., Critelli, P., Chillari, F., Ferrantelli, G., Sciacca, M., Brogna, A., Parisi, S., & Pergolizzi, S. (2023). Modulation of Radiation Doses and Chimeric Antigen Receptor T Cells: A Promising New Weapon in Solid Tumors—A Narrative Review. Journal of Personalized Medicine, 13(8), 1261. https://doi.org/10.3390/jpm13081261