

Atypical Foveal Hypoplasia in Best Disease

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Participants and Data Collection
2.3. B-Scan SD-OCT Analysis
2.4. OCT-A Analysis
2.5. Statistical Analysis
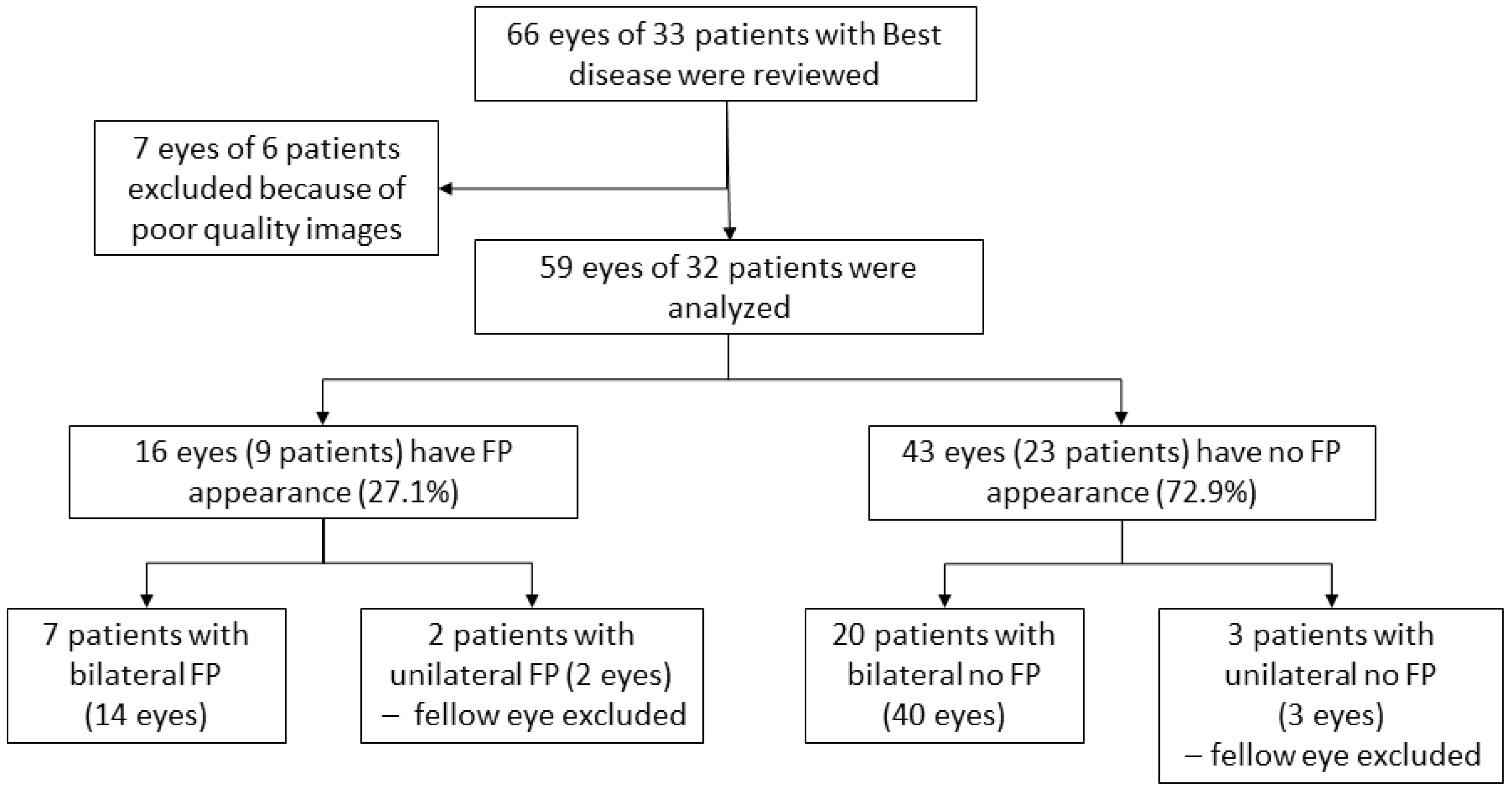
3. Results
3.1. Patients Characteristics
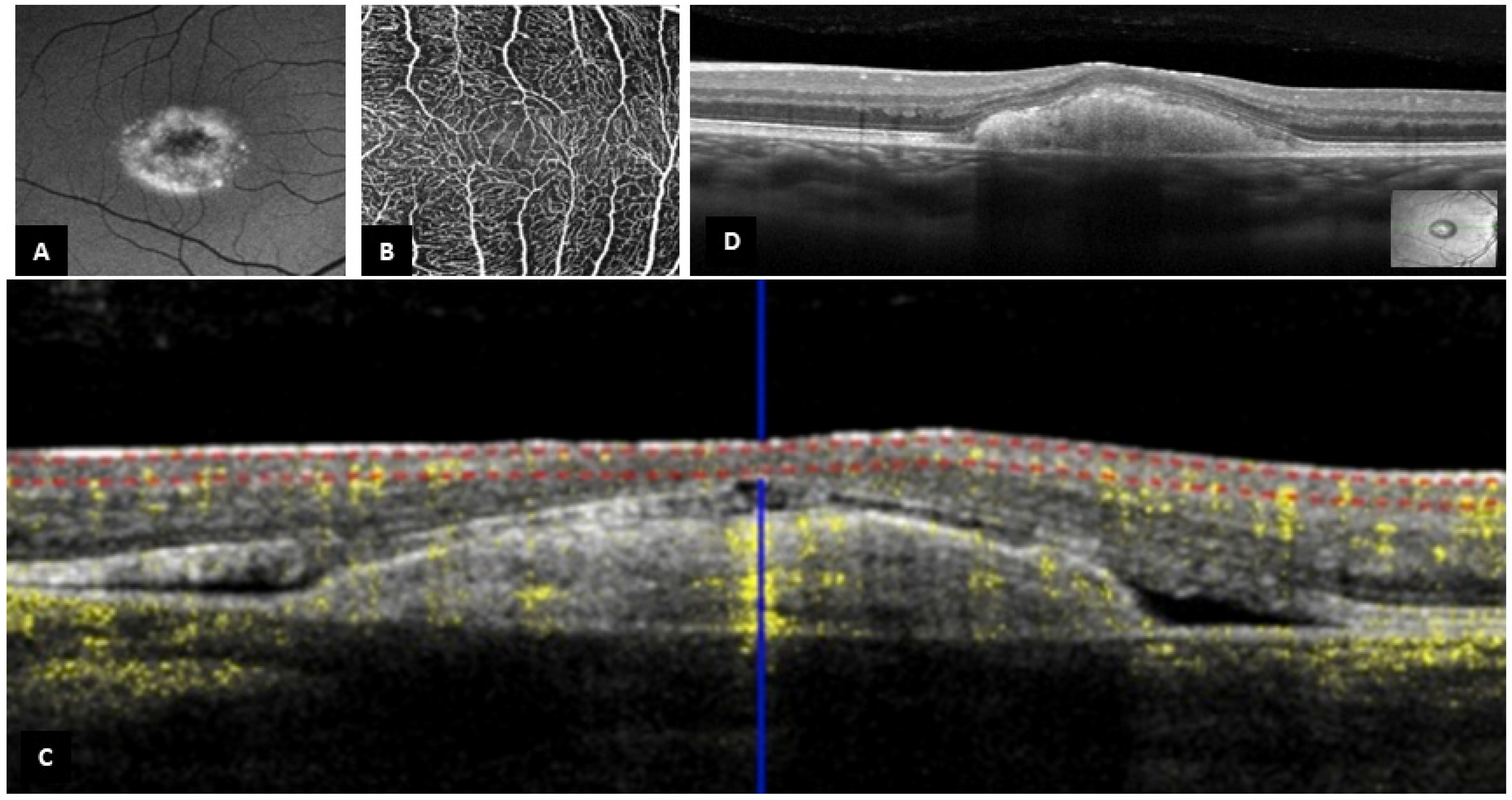
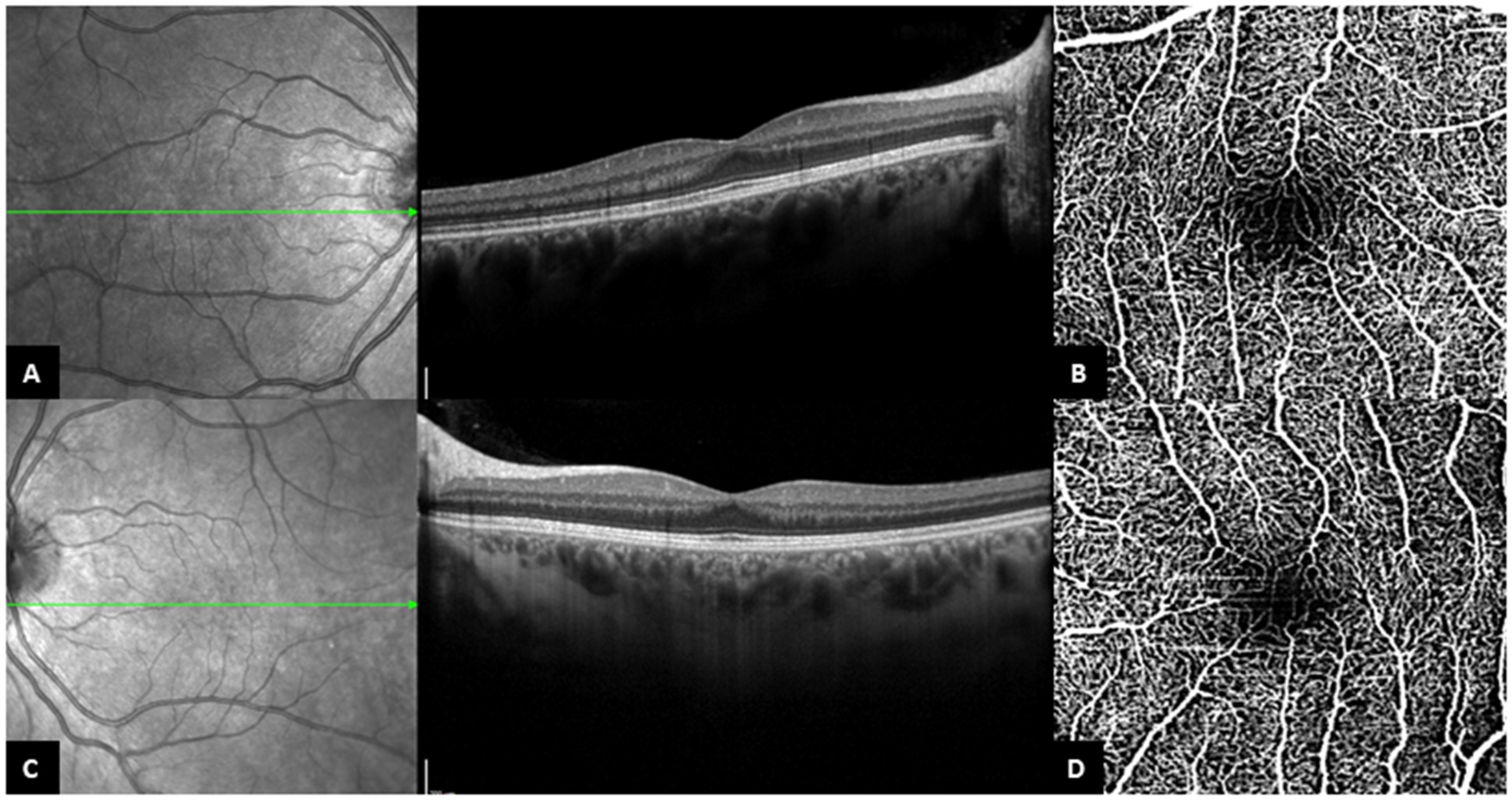
3.2. B-Scan SD-OCT Findings
3.3. OCT-A Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tripathy, K.; Salini, B. Best Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sun, H.; Tsunenari, T.; Yau, K.-W.; Nathans, J. The Vitelliform Macular Dystrophy Protein Defines a New Family of Chloride Channels. Proc. Natl. Acad. Sci. USA 2002, 99, 4008–4013. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, H.C.; Qu, Z.; Yu, K.; Xiao, Q.; Chien, L.-T. Molecular Physiology of Bestrophins: Multifunctional Membrane Proteins Linked to Best Disease and Other Retinopathies. Physiol. Rev. 2008, 88, 639–672. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the Product of the Best Vitelliform Macular Dystrophy Gene (VMD2), Localizes to the Basolateral Plasma Membrane of the Retinal Pigment Epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.X.; Charran, D.; Strong, C.R.; Steffens, T.J.; Jayasundera, T.; Heckenlively, J.R. Optical Coherence Tomography Examination of the Retinal Pigment Epithelium in Best Vitelliform Macular Dystrophy. Ophthalmology 2017, 124, 456–463. [Google Scholar] [CrossRef]
- Creel, D.J. The Electrooculogram. Handb. Clin. Neurol. 2019, 160, 495–499. [Google Scholar]
- Constable, P.A.; Bach, M.; Frishman, L.J.; Jeffrey, B.G.; Robson, A.G. International Society for Clinical Electrophysiology of Vision ISCEV Standard for Clinical Electro-Oculography (2017 Update). Doc. Ophthalmol. 2017, 134, 1–9. [Google Scholar] [CrossRef]
- Spaide, R.F.; Noble, K.; Morgan, A.; Freund, K.B. Vitelliform Macular Dystrophy. Ophthalmology 2006, 113, 1392–1400. [Google Scholar] [CrossRef]
- Ferrara, D.C.; Costa, R.A.; Tsang, S.; Calucci, D.; Jorge, R.; Freund, K.B. Multimodal Fundus Imaging in Best Vitelliform Macular Dystrophy. Graefes Arch. Clin. Exp. Ophthalmol. 2010, 248, 1377–1386. [Google Scholar] [CrossRef]
- Guduru, A.; Gupta, A.; Tyagi, M.; Jalali, S.; Chhablani, J. Optical Coherence Tomography Angiography Characterisation of Best Disease and Associated Choroidal Neovascularisation. Br. J. Ophthalmol. 2018, 102, 444–447. [Google Scholar] [CrossRef]
- Mirshahi, A.; Lashay, A.; Masoumi, A.; Abrishami, M. Optical Coherence Tomography Angiography in Best Vitelliform Macular Dystrophy. J. Curr. Ophthalmol. 2019, 31, 442–445. [Google Scholar] [CrossRef]
- Marmor, M.F.; Choi, S.S.; Zawadzki, R.J.; Werner, J.S. Visual Insignificance of the Foveal Pit: Reassessment of Foveal Hypoplasia as Fovea Plana. Arch. Ophthalmol. 2008, 126, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Chatzistergiou, V.; Cilliers, H.; Pournaras, J.-A.; Ambresin, A. Fovea Plana On Optical Coherence Tomography Angiography: New Perspectives. Retina 2021, 41, 1541–1546. [Google Scholar] [CrossRef]
- Thomas, M.G.; Papageorgiou, E.; Kuht, H.J.; Gottlob, I. Normal and Abnormal Foveal Development. Br. J. Ophthalmol. 2020, 106, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Knight, D.; Nudleman, E. Multimodal Imaging of Idiopathic Fovea Plana. Ophthalmol. Retina 2020, 4, 972. [Google Scholar] [CrossRef]
- Singh, S.R.; Jain, S.; Kaur, S.; Dogra, M. Multimodal Imaging of Fovea Plana in Oculocutaneous Albinism. Indian J. Ophthalmol. 2019, 67, 1733. [Google Scholar]
- Noval, S.; Freedman, S.F.; Asrani, S.; El-Dairi, M.A. Incidence of Fovea Plana in Normal Children. J. AAPOS 2014, 18, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Kumar, A.; Mohammad, S.; Proudlock, F.A.; Engle, E.C.; Andrews, C.; Chan, W.-M.; Thomas, S.; Gottlob, I. Structural Grading of Foveal Hypoplasia Using Spectral-Domain Optical Coherence Tomography a Predictor of Visual Acuity? Ophthalmology 2011, 118, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F. The Ambiguity of Pachychoroid. Retina 2021, 41, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Lejoyeux, R.; Bonnin, S.; Guindolet, D.; Jacquiod, B.; Erol, O.; Le Mer, Y.; Jeguirim, H.; Mauget-Faÿsse, M.; Tadayoni, R. Incidence and Clinical Significance of Fovea Plana in the French Population with Age-Related Cataract. Eur. J. Ophthalmol. 2022; ahead-of-print. [Google Scholar] [CrossRef]
- Hendrickson, A.E.; Yuodelis, C. The Morphological Development of the Human Fovea. Ophthalmology 1984, 91, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Yuodelis, C.; Hendrickson, A. A Qualitative and Quantitative Analysis of the Human Fovea during Development. Vision Res. 1986, 26, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Provis, J.M.; Hendrickson, A.E. The Foveal Avascular Region of Developing Human Retina. Arch. Ophthalmol. 2008, 126, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Zobor, D.; Chiang, W.-C.; Weisschuh, N.; Staller, J.; Gonzalez Menendez, I.; Chang, S.; Beck, S.C.; Garcia Garrido, M.; Sothilingam, V.; et al. Mutations in the Unfolded Protein Response Regulator ATF6 Cause the Cone Dysfunction Disorder Achromatopsia. Nat. Genet. 2015, 47, 757–765. [Google Scholar] [CrossRef]
- Skorczyk-Werner, A.; Chiang, W.-C.; Wawrocka, A.; Wicher, K.; Jarmuż-Szymczak, M.; Kostrzewska-Poczekaj, M.; Jamsheer, A.; Płoski, R.; Rydzanicz, M.; Pojda-Wilczek, D.; et al. Autosomal Recessive Cone-Rod Dystrophy Can Be Caused by Mutations in the ATF6 Gene. Eur. J. Hum. Genet. 2017, 25, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, H.; Grandjean, J.M.D.; Chiang, W.-C.J.; Bindels, D.D.; Mastey, R.; Okalova, J.; Nguyen, A.; Powers, E.T.; Kelly, J.W.; Grimsey, N.J.; et al. ATF6 Is Essential for Human Cone Photoreceptor Development. Proc. Natl. Acad. Sci. USA 2021, 118, e2103196118. [Google Scholar] [CrossRef]
- Milenkovic, A.; Milenkovic, V.M.; Wetzel, C.H.; Weber, B.H.F. BEST1 Protein Stability and Degradation Pathways Differ between Autosomal Dominant Best Disease and Autosomal Recessive Bestrophinopathy Accounting for the Distinct Retinal Phenotypes. Hum. Mol. Genet. 2018, 27, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Batıoğlu, F.; Yanık, Ö.; Demirel, S.; Çağlar, Ç.; Özmert, E. A Case of Best Disease Accompanied by Pachychoroid Neovasculopathy. Turk. J. Ophthalmol 2019, 49, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Samara, W.A.; Say, E.A.T.; Khoo, C.T.L.; Higgins, T.P.; Magrath, G.; Ferenczy, S.; Shields, C.L. Correlation of foveal avascular zone size with foveal morphology in normal eyes using optical coherence tomography angiography. Retina 2015, 35, 2188–2195. [Google Scholar] [CrossRef]
- Battaglia Parodi, M.; Romano, F.; Cicinelli, M.V.; Rabiolo, A.; Arrigo, A.; Pierro, L.; Iacono, P.; Bandello, F. Retinal Vascular Impairment in Best Vitelliform Macular Dystrophy Assessed by Means of Optical Coherence Tomography Angiography. Am. J. Ophthalmol. 2018, 187, 61–70. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patient | Age (yrs) | Sex | R/L | SE (D) | VA (logMAR) | IOP | FP Y/N/E | FP Grade | Diagnose Made Based on | BEST1 Mutation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7 | F | R L | - - | 0.1 0.1 | - - | Y E | Atypical - | Familial history and clinical examination | - |
| 2 FP | 7 | F | R L | +1.875 +1.625 | 0.0 0.1 | 16 16 | Y Y | Atypical Atypical | Familial history, clinical examination, EOG Arden ration less than 170%, genetic confirmation | het. mutation c.44G>A p.(Gly15Asp) exon 2 |
| 3 | 26 | F | R L | +3.25 +1.50 | 0.7 0.18 | 14 15 | N N | - - | Clinical examination, EOG Arden ration less than 170%, genetic confirmation | mutation het.c.44G>A, p.(Gly15Asp) exon 2 |
| 4 | 78 | F | R L | - - | 0.9 0.9 | 11 12 | N N | - - | Familial history and clinical examination | - |
| 5 | 55 | M | R L | +4.0 +3.625 | 0.152 0.3 | 14 14 | N N | - - | Familial history, clinical examination, EOG Arden ration less than 170% | - |
| 6 | 35 | M | R L | +0.125 +0.625 | 0.0 0.0 | 11 9 | N N | - - | Familial history, clinical examination, genetic confirmation | het. mutation c.905A>C (Asp302Ala) |
| 7 | 15 | M | R L | +0.125 +1.0 | −0.10 0.0 | 17 18 | Y Y | Atypical Atypical | Clinical examination alone | - |
| 8 | 34 | F | R L | −1.625 −1.625 | 0.3 0.3 | 11 14 | N N | - - | Clinical examination alone | - |
| 9 | 62 | F | R L | - - | 0.045 0.045 | 16 14 | N N | - - | Familial history, clinical examination, EOG Arden ration 170–179%, genetic confirmation | het. mutation c.97T>C, p.(Tyr33His) exon 2 |
| 10 | 37 | F | R L | +0.125 +0.125 | 0.4 0.2 | 18 13 | N N | - - | Clinical examination alone | - |
| 11 | 57 | F | R L | +3.0 +2.325 | 0.3 0.6 | 15 13 | E E | - - | Familial history, clinical examination, EOG Arden ration less than 170% | - |
| 12 | 29 | F | R L | −0.50 −2.25 | 0.3 0.3 | 14 17 | N N | - - | Familial history, clinical examination, EOG Arden ration less than 170%, genetic confirmation | het. mutation c.916G>C, p.(Glu306Gln) exon 8 |
| 13 | 11 | F | R L | +1.375 +1.625 | 0.0 0.0 | 16 16 | N N | - - | Clinical examination, EOG Arden ration 170–179% | - |
| 14 | 45 | M | R L | +1.125 +1.75 | 0.3 0.2 | 13 15 | N N | - - | Familial history, clinical examination, EOG Arden ration less than 170%, genetic confirmation | het. mutation c.10A>G (p.Thr4Ala) exon 2 |
| 15 | 6 | F | R L | +5.50 +0.125 | 0.152 0.7 | 17 17 | Y E | Atypical - | Clinical examination and genetic confirmation | hom. mutation (recessive) IVS5+ 1G>A |
| 16 | 45 | F | R L | - - | 0.2 0.2 | 13 14 | N N | - - | Clinical examination, EOG Arden ration less than 170% | - |
| 17 | 58 | M | R L | +5.625 +5.75 | 0.3 1.0 | 18 18 | N N | - - | Familial history, clinical examination, genetic confirmation | het. mutation c.692G>C, p.(Ser231Thr) exon 6 |
| 18 | 29 | F | R L | −0.125 −0.625 | 0.0 0.0 | 13 15 | Y Y | 1b 1b | Familial history and genetic confirmation | het. mutation c.692G>C, p.(Ser231Thr) exon 6 |
| 19 | 19 | F | R L | +2.125 +1.375 | 0.3 0.5 | 15 16 | N N | - - | Familial history and clinical examination | - |
| 20 | 11 | M | R L | +1.625 +1.50 | 0.3 0.4 | 12 15 | Y Y | Atypical Atypical | Familial history and clinical examination | - |
| 21 | 15 | M | R L | - - | 0.4 0.7 | - - | E N | - - | Familial history, clinical examination, genetic confirmation | het. mutation c.25G>T, p.(Val9Leu) exon 2 |
| 22 | 12 | M | R L | +0.25 +3.75 | 0.5 0.0 | 17 18 | N N | - - | Familial history, clinical examination, genetic confirmation | het. mutation c.25G>T, p.(Val9Leu) exon 2 |
| 23 | 10 | F | R L | +0.50 +0.625 | 0.0 0.0 | - - | N N | - - | Familial history and clinical examination | - |
| 24 | 14 | M | R L | - - | 0.0 0.1 | - - | N N | - - | Familial history and clinical examination | - |
| 25 | 37 | M | R L | - - | 0.4 0.1 | - - | E N | - - | Familial history, clinical examination, genetic confirmation | het. mutation c.217A>T, p.(Ile73Phe) exon 3 |
| 26 | 37 | M | R L | +6.0 +4.625 | 1.30 0.2 | 13 16 | E N | - - | Familial history, clinical examination, EOG Arden ration less than 170%, genetic confirmation | het. mutation c.916G>C, p.(Glu306Gln) exon 8 |
| 27 | 14 | M | R L | −0.125 +1.25 | 0.4 0.0 | 20 19 | Y Y | Atypical Atypical | Familial history and clinical examination | - |
| 28 | 16 | M | R L | +5.50 +6.0 | 0.0 0.6 | 20 21 | N N | - - | Familial history and clinical examination | - |
| 29 | 49 | M | R L | +4.75 +5.375 | 0.20 1.1 | 19 20 | N N | - - | Familial history and clinical examination | - |
| 30 | 47 | M | R L | +0.25 +0.125 | 0.5 0.7 | 12 12 | Y Y | Atypical Atypical | Clinical examination alone | - |
| 31 | 31 | M | R L | +0.125 0 | 0.0 0.0 | 12 15 | N N | - - | Clinical examination and EOG Arden ration less than 170% | - |
| 32 | 60 | F | R L | - - | −0.10 1.1 | 14 15 | N N | - - | Familial history and clinical examination | - |
| 33 | 31 | M | R L | −2.25 −2.25 | 0.4 0.0 | 17 18 | Y Y | Atypical Atypical | Clinical examination alone | - |
| FP Group (n = 9) | No FP Group (n = 23) | All (n = 32) | p-Value | |
|---|---|---|---|---|
| Gender Male (%) Female (%) | 5 (55.6%) 4 (44.4%) | 12 (52.2%) 11 (47.8%) | 17 (53.1%) 15 (46.9%) | NA |
| Age (years) Mean (SD) Median (Q1, Q3) Min-Max | 19.17 (14.10) 14.70 (7.40, 29.80) 6.70–47.30 | 35.92 (18.87) 35.40 (18.20, 47.45) 10.10–78.10 | 31.21 (19.04) 30.85 (14.57, 45.32) 6.70–78.10 | 0.01 † |
| FP Group (n = 16) | No FP Group (n = 43) | All (n = 59) | p-Value | |
|---|---|---|---|---|
| Eyes RE (%) LE (%) | 9 (56.2%) 7 (43.8%) | 20 (46.5%) 23 (53.5%) | 29 (49.2%) 30 (50.8%) | 0.5 * |
| Visual acuity (logMAR) Mean (SD) Median (Q1, Q3) Min-Max | 0.18 (0.23) 0.10 (0.00, 0.40) −0.10–0.70 | 0.29 (0.33) 0.20 (0.00, 0.35) −0.10–1.10 | 0.26 (0.30) 0.20 (0.00, 0.40) −0.10–1.10 | 0.3 † |
| IOP Mean (SD) Median (Q1, Q3) Min-Max Missing | 15.80 (2.60) 16.00 (14.00, 17.50) 12.00–20.00 1 | 15.08 (2.75) 15.00 (14.00, 17.00) 9.00–21.00 6 | 15.29 (2.70) 15.00 (13.75, 17.00) 9.00–21.00 7 | 0.4 ‡ |
| Spherical equivalent (phakic eyes only) (D) Mean (SD) Median (Q1, Q3) Min-Max Missing | 0.63 (1.86) 0.25 (−0.12, 1.56) −2.25–5.50 1 | 1.92 (2.40) 1.38 (0.12, 3.88) −2.25–6.00 3 | 1.50 (2.30) 1.19 (0.12, 2.97) −2.25–6.00 4 | 0.05 ‡ |
| Stage of the disease 1 2 3 4 5 6 | 2 (12.5%) 0 (0.0%) 3 (18.8%) 3 (18.8%) 0 (0.0%) 8 (50.0%) | 3 (7.0%) 6 (14.0%) 7 (16.3%) 2 (4.7%) 4 (9.3%) 21 (48.8%) | 5 (8.5%) 6 (10.2%) 10 (16.9%) 5 (8.5%) 4 (6.8%) 29 (49.2%) | 0.3 ** |
| Persistence of vitelliform material under the macula No (%) Yes (%) | 2 (12.5%) 14 (87.5%) | 8 (18.6%) 35 (81.4%) | 10 (16.9%) 49 (83.1%) | 0.7 ** |
| Choroidal thickness (µm) Mean (SD) Median (Q1, Q3) Min-Max Missing | 338.25 (92.20) 360.00 (306.50, 395.50) 92.00–445.00 0 | 360.52 (102.71) 364.50 (319.75, 404.00) 118.00–613.00 3 | 354.16 (99.51) 364.50 (311.75, 398.50) 92.00–613.00 3 | 0.7 † |
| Presence of pachyvessels No (%) Yes (%) | 4 (25.0%) 12 (75.0%) | 8 (18.6%) 35 (81.4%) | 12 (20.3%) 47 (79.7%) | 0.7 ** |
| Stage I (n = 5) | Stage II (n = 6) | Stage III (n = 10) | Stage IV (n = 5) | Stage V (n = 4) | Stage VI (n = 29) | p-Value | |
|---|---|---|---|---|---|---|---|
| Choroidal thickness (µm) | 0.7 * | ||||||
| Mean (SD) | 374.20 (54.73) | 394.40 (88.62) | 350.00 (40.49) | 353.00 (85.85) | 302.25 (74.08) | 352.44 (127.01) | |
| Median (Q1, Q3) | 394.00 (352.00, 397.00) | 361.00 (339.00, 435.00) | 334.50 (320.25, 385.50) | 397.00 (292.00, 417.00) | 313.00 (258.25, 357.00) | 368.00 (311.50, 401.50) | |
| Min-Max | 292.00–436.00 0 | 308.00–529.00 | 293.00–407.00 | 233.00–426.00 | 211.00–372.00 | 92.00–613.00 | |
| Missing | 374.20 (54.73) | 1 | 0 | 0 | 2 | ||
| Pachyvessels | 0.4 † | ||||||
| No (%) | 1 (20.0%) | 1 (16.7%) | 2 (20.0%) | 2 (40.0%) | 2 (50.0%) | 4 (13.8%) | |
| Yes (%) | 4 (80.0%) | 5 (83.3%) | 8 (80.0%) | 3 (60.0%) | 2 (50.0%) | 25 (86.2%) |
| FP Group (n = 13) | No FP Group (n = 27) | All (n = 40) | p-Value | |
|---|---|---|---|---|
| Bridging vessels across de fovea | <0.001 * | |||
| No (%) | 0 (0%) | 27 (100%) | 27 (67.5%) | |
| Yes (%) | 13 (100%) | 0 (0%) | 13 (32.5%) | |
| FAZ area (mm2) | 0.34 ± 0.17 mm2 |
| Stage I (n = 5) | Stage II (n = 6) | Stage III (n = 10) | Stage IV (n = 5) | Stage V (n = 4) | Stage VI (n = 29) | p-Value | |
|---|---|---|---|---|---|---|---|
| FAZ area (mm2) | 0.2 * | ||||||
| Mean (SD) | 0.41 (0.01) | 0.29 (0.03) | 0.34 (0.14) | 0.20 (0.13) | 0.45 (0.07) | 0.36 (0.23) | |
| Median (Q1, Q3) | 0.41 (0.41, 0.42) | 0.29 (0.28, 0.31) | 0.38 (0.31, 0.44) | 0.20 (0.15, 0.24) | 0.45 (0.42, 0.47) | 0.30 (0.24, 0.36) | |
| Min-Max | 0.41–0.42 | 0.25–0.33 | 0.10–0.46 | 0.11–0.29 | 0.40–0.50 | 0.19–1.00 | |
| Missing | 3 | 2 | 4 | 3 | 2 | 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moret, E.; Lejoyeux, R.; Bonnin, S.; Azar, G.; Guillaume, J.; Le Cossec, C.; Lafolie, J.; Alonso, A.-S.; Favard, C.; Meunier, I.; et al. Atypical Foveal Hypoplasia in Best Disease. J. Pers. Med. 2023, 13, 337. https://doi.org/10.3390/jpm13020337
Moret E, Lejoyeux R, Bonnin S, Azar G, Guillaume J, Le Cossec C, Lafolie J, Alonso A-S, Favard C, Meunier I, et al. Atypical Foveal Hypoplasia in Best Disease. Journal of Personalized Medicine. 2023; 13(2):337. https://doi.org/10.3390/jpm13020337
Chicago/Turabian StyleMoret, Emmanuelle, Raphaël Lejoyeux, Sophie Bonnin, Georges Azar, Jessica Guillaume, Chloé Le Cossec, Justine Lafolie, Anne-Sophie Alonso, Catherine Favard, Isabelle Meunier, and et al. 2023. "Atypical Foveal Hypoplasia in Best Disease" Journal of Personalized Medicine 13, no. 2: 337. https://doi.org/10.3390/jpm13020337
APA StyleMoret, E., Lejoyeux, R., Bonnin, S., Azar, G., Guillaume, J., Le Cossec, C., Lafolie, J., Alonso, A.-S., Favard, C., Meunier, I., Vasseur, V., & Mauget-Faÿsse, M. (2023). Atypical Foveal Hypoplasia in Best Disease. Journal of Personalized Medicine, 13(2), 337. https://doi.org/10.3390/jpm13020337