

Comprehensive Analysis of Necroptosis Landscape in Skin Cutaneous Melanoma for Appealing its Implications in Prognosis Estimation and Microenvironment Status

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Online Data
2.2. Clinical Tissue Specimens
2.3. Analysis of Differentially NRGs
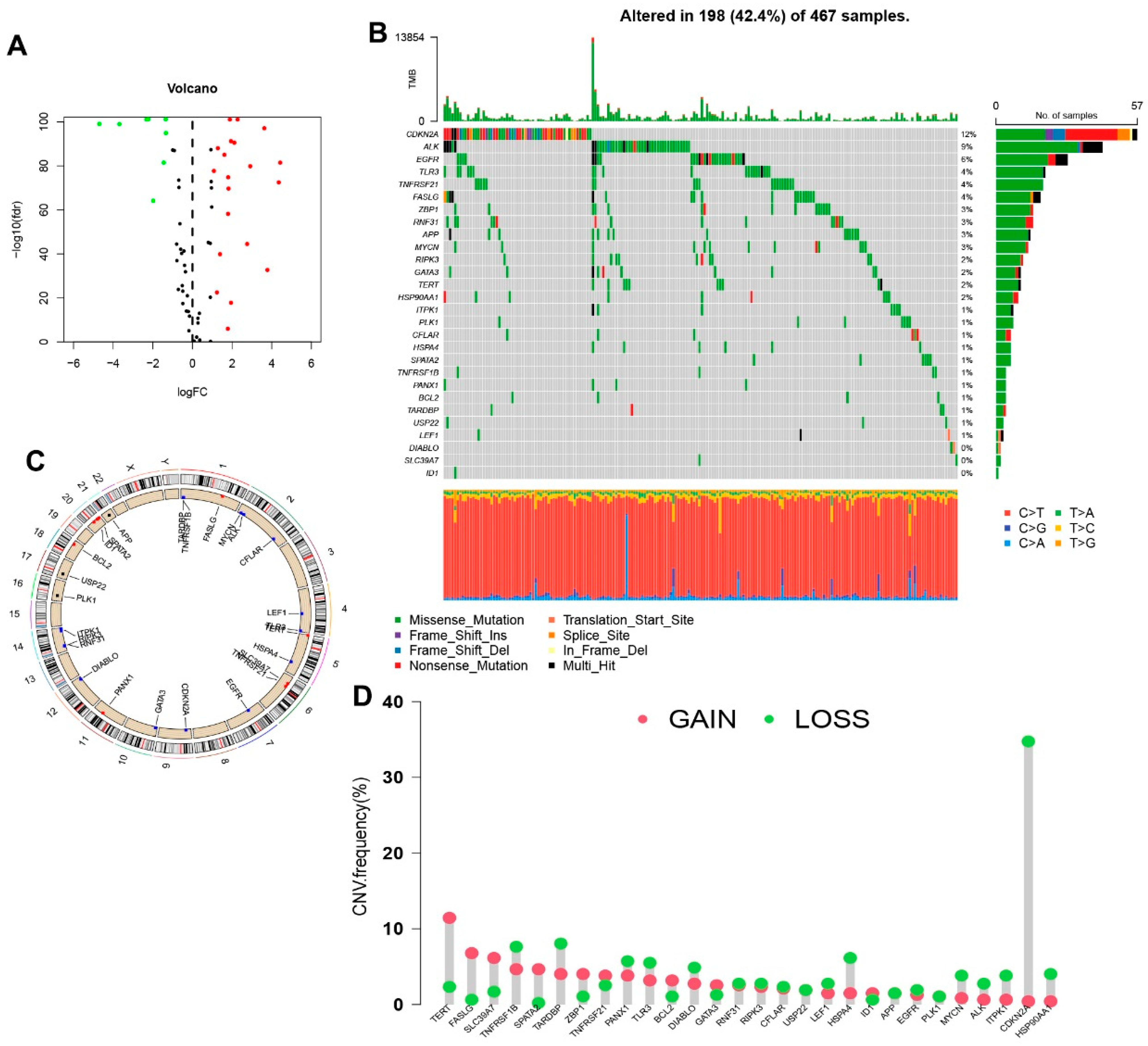
2.4. Genetic Mutation and Expression Variation Analysis
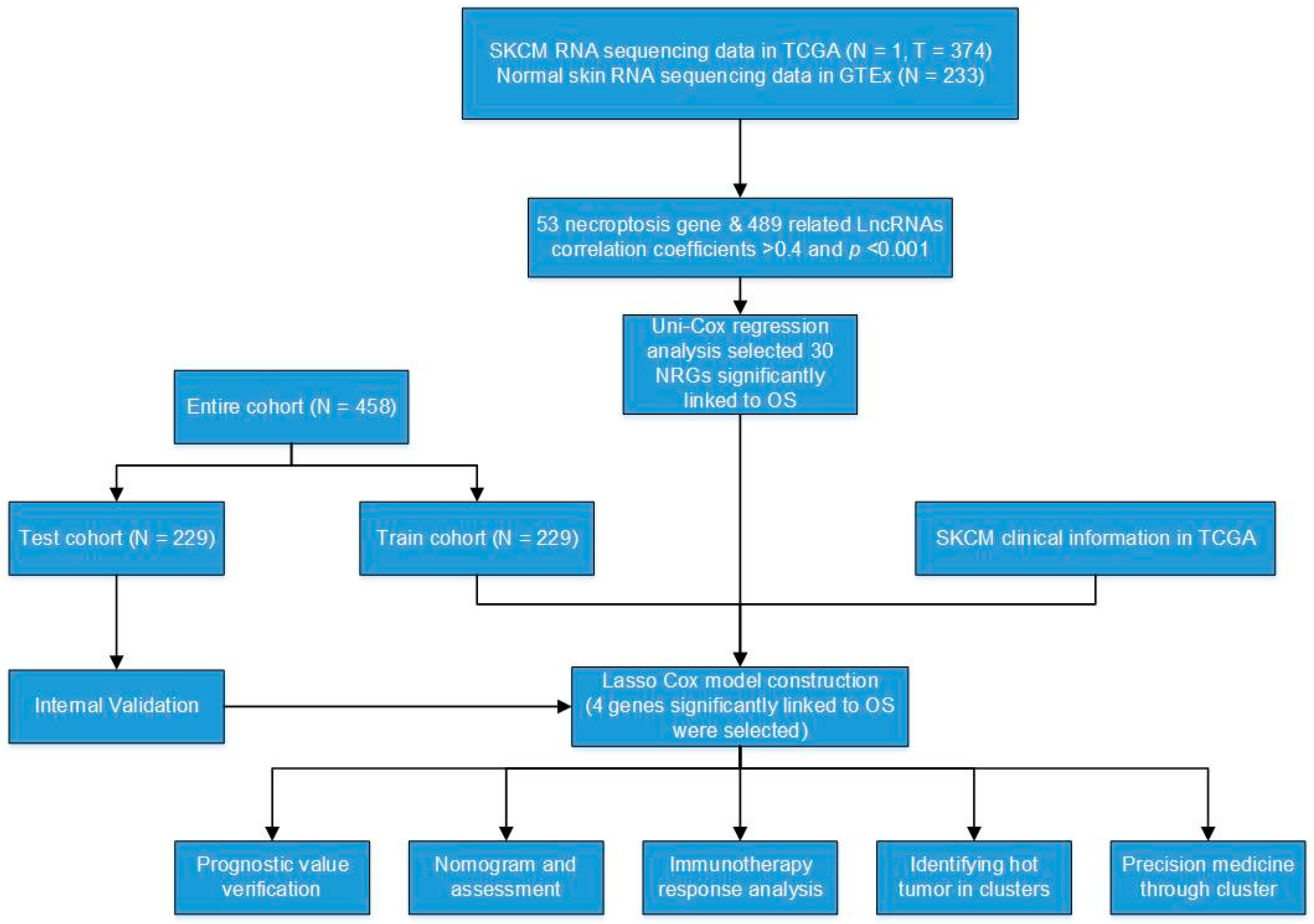
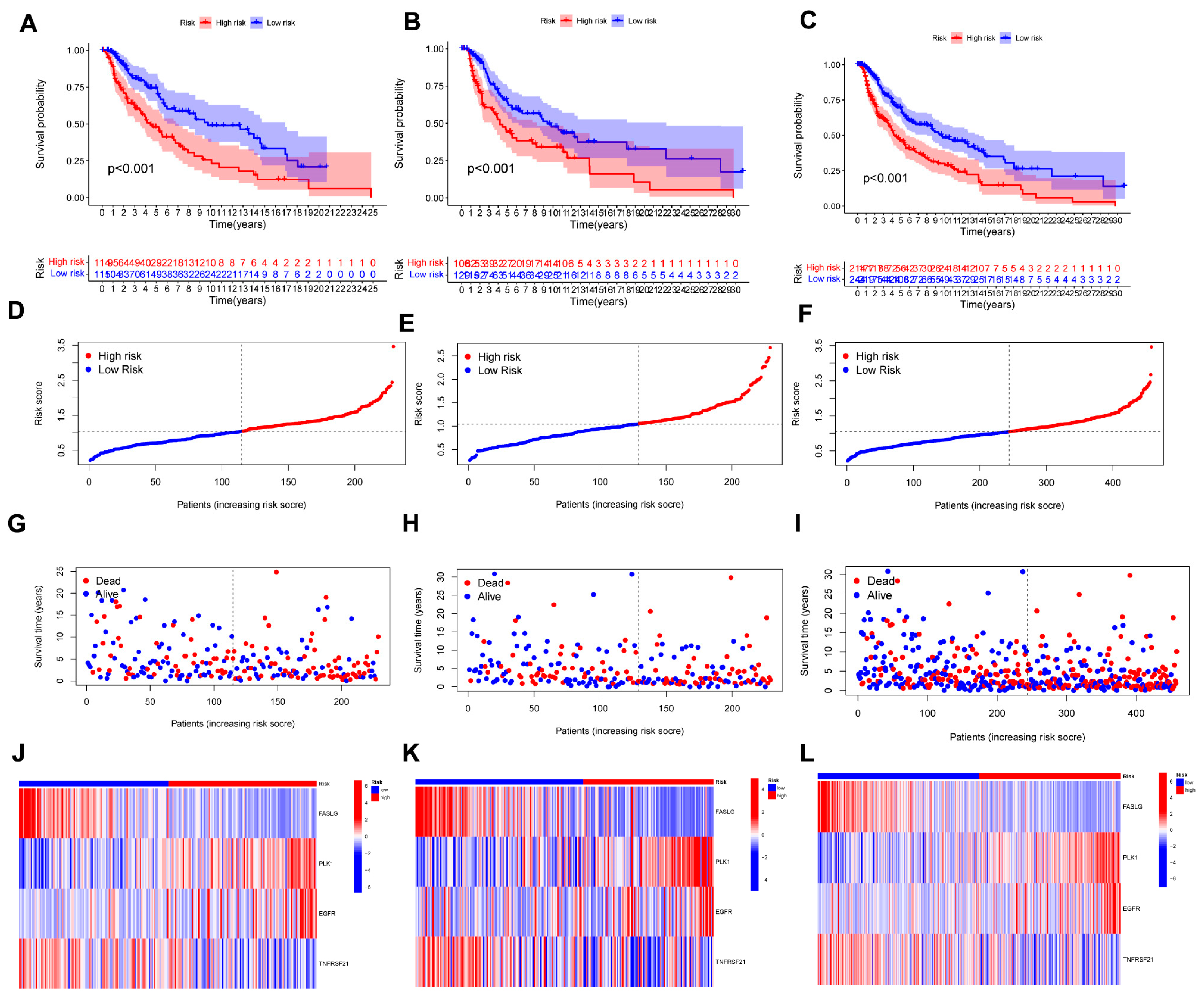
2.5. Construction and Validation of the necroptosis-Related Signature
2.6. Independence Factors and ROC
2.7. Nomogram and Calibration
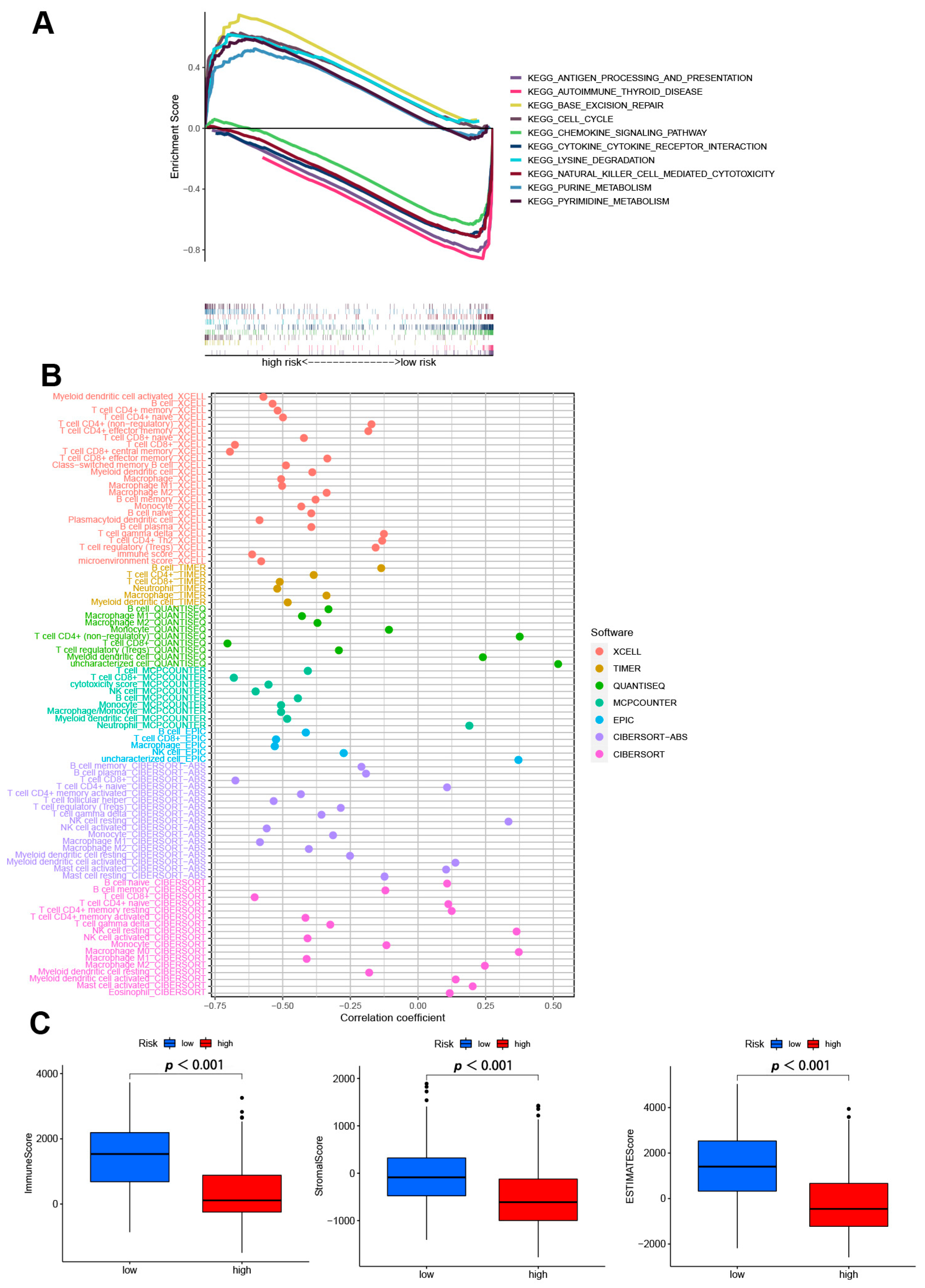
2.8. Gene Set Enrichment Analyses
2.9. TME and Immune Checkpoints Analyses
2.10. Potential Therapeutic Medicine
2.11. Clusters Based on Four Prognostic Signature Genes
2.12. Immunohistochemical Staining (IHC)
3. Results
3.1. The Genetic Mutation Landscape and Expression of Necroptosis Genes in SKCM
3.2. Construction and Validation of a Necroptosis-Related Prognostic Signature
3.3. Nomogram Analysis
3.4. Evaluation of the Risk Signature
3.5. GSEA Analyses
3.6. Analysis of Immune Factors and Clinical Treatment in Risk Groups
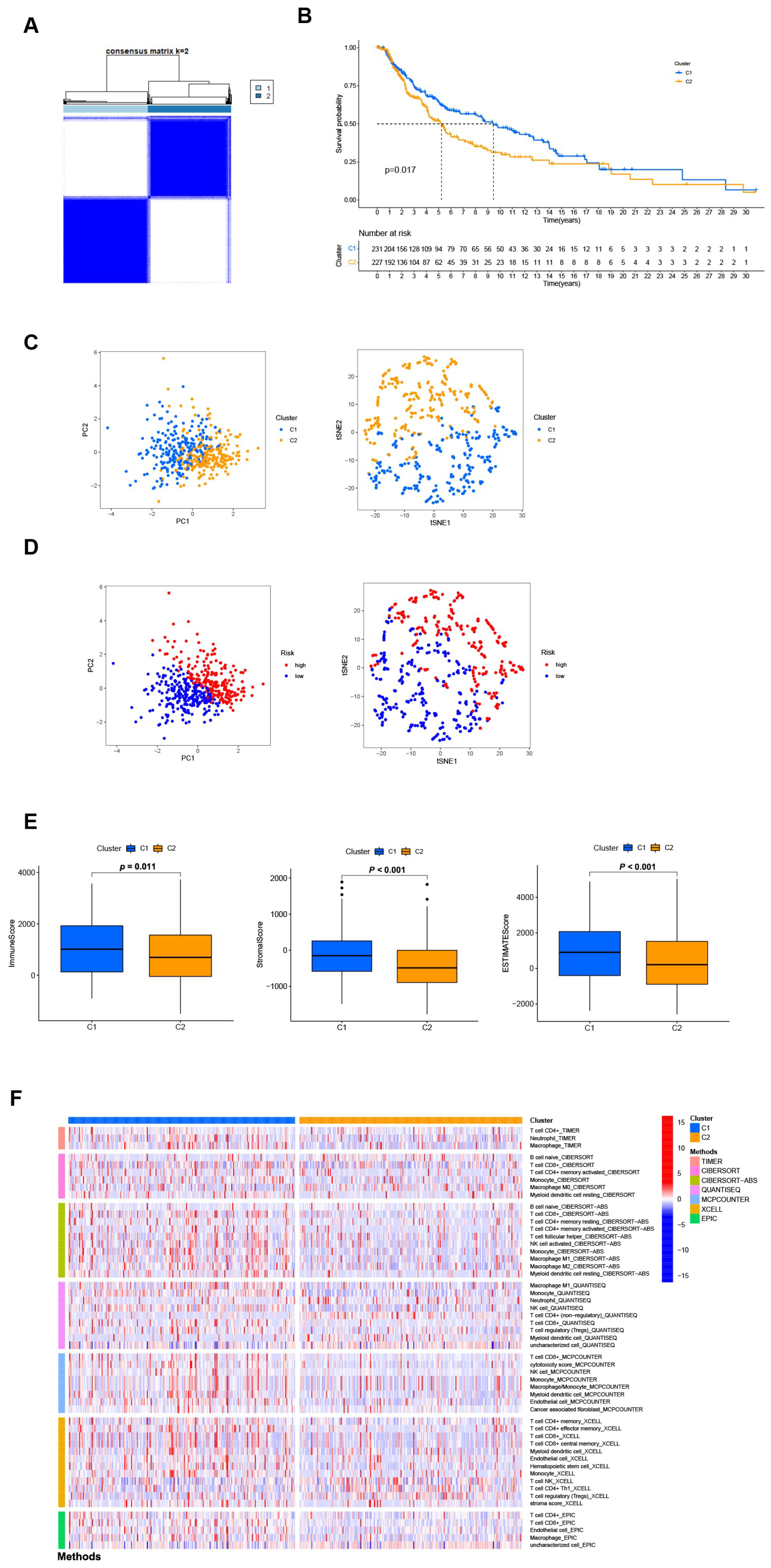
3.7. Distinguishing between the Different Subtypes and Precision Medicine in Clusters
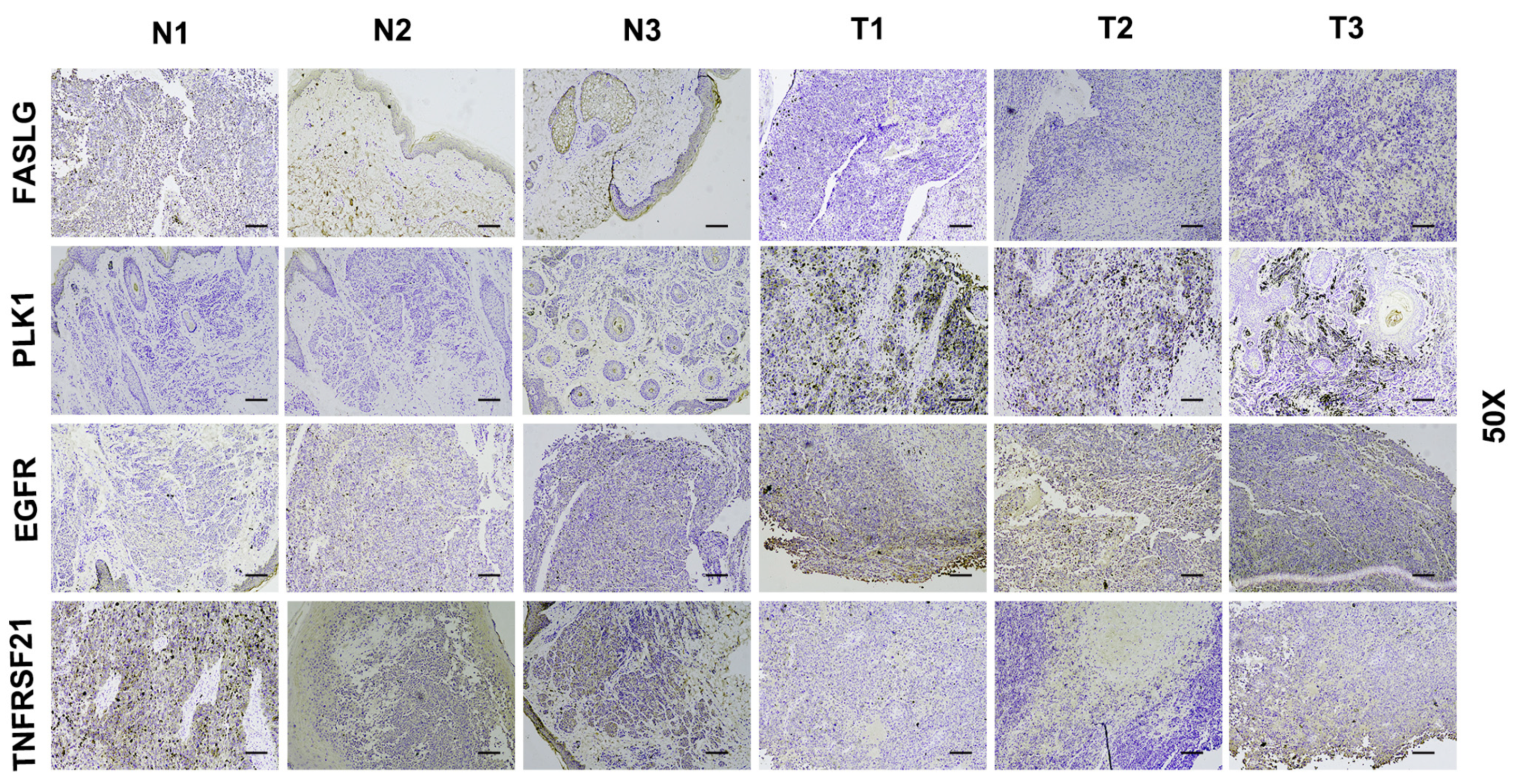
3.8. Verify Signature Gene Expression by IHC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Ossio, R.; Roldán-Marín, R.; Martínez-Said, H.; Adams, D.J.; Espinoza, C.D.R. Melanoma: A global perspective. Nat. Rev. Cancer 2017, 17, 393–394. [Google Scholar] [CrossRef]
- Lattanzi, M.; Lee, Y.; Simpson, D.; Moran, U.; Darvishian, F.; Kim, R.; Hernando, E.; Polsky, D.; Hanniford, D.; Shapiro, R.; et al. Primary Melanoma Histologic Subtype: Impact on Survival and Response to Therapy. J. Natl. Cancer Inst. 2019, 111, 180–188. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, Q.; Wang, Y.; Zhang, L.; Zhao, H.; Chen, C.; Wang, Y.; Liu, S.; Lu, T.; Wang, F.; et al. Molecular classification and subtype-specific characterization of skin cutaneous melanoma by aggregating multiple genomic platform data. J. Cancer Res. Clin. Oncol. 2018, 144, 1635–1647. [Google Scholar] [CrossRef]
- Zhang, W.; Kong, Y.; Li, Y.; Shi, F.; Lyu, J.; Sheng, C.; Wang, S.; Wang, Q. Novel Molecular Determinants of Response or Resistance to Immune Checkpoint Inhibitor Therapies in Melanoma. Front Immunol. 2022, 12, 798474. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Orozco, S.; Oberst, A. RIPK3 in cell death and inflammation: The good, the bad, and the ugly. Immunol. Rev. 2017, 277, 102–112. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Li, Y.; Yuan, J. Control of life-or-death decisions by RIP1 kinase. Annu. Rev. Physiol. 2014, 76, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Ly, N.N.G.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Therapeutic exploitation of necroptosis for cancer therapy. Semin. Cell Dev. Biol. 2014, 35, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Negroni, A.; Colantoni, E.; Cucchiara, S.; Stronati, L. Necroptosis in Intestinal Inflammation and Cancer: New Concepts and Therapeutic Perspectives. Biomolecules 2020, 10, 1431. [Google Scholar] [CrossRef]
- Kong, Q.; Lv, J.; Yan, S.; Chang, K.-J.; Wang, G. A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells. Int. J. Mol. Sci. 2018, 19, 2975. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Seo, J.; Nam, Y.W.; Kim, S.; Oh, D.-B.; Song, J. Necroptosis molecular mechanisms: Recent findings regarding novel necroptosis regulators. Exp. Mol. Med. 2021, 53, 1007–1017. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Zhou, X.; Fang, D.; Ou, X.; Ye, J.; Peng, J.; Xu, J. Necroptosis-Related lncRNAs: Predicting Prognosis and the Distinction between the Cold and Hot Tumors in Gastric Cancer. J. Oncol. 2021, 2021, 6718443. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J. Tumor Microenvironment, Metabolism, and Immunotherapy. N. Engl. J. Med. 2020, 382, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; Van Oppen, L.M.P.E.; Schöckel, L.; Bossenbroek, H.M.; Van Emst-de Vries, S.E.; Hermeling, J.C.W.; Grefte, S.; Kopitz, C.; Heroult, M.; Willems, P.H.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Raes, L.; Stremersch, S.; Brans, T.; Fraire, J.C.; Roelandt, R.; Declercq, W.; Vandenabeele, P.; Raemdonck, K.; Braeckmans, K.; et al. Delivery of Mixed-Lineage Kinase Domain-Like Protein by Vapor Nanobubble Photoporation Induces Necroptotic-Like Cell Death in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 4254. [Google Scholar] [CrossRef]
- Hou, J.; Ju, J.; Zhang, Z.; Zhao, C.; Li, Z.; Zheng, J.; Sheng, T.; Zhang, H.; Hu, L.; Yu, X.; et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 2019, 10, 493. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef] [PubMed]
- Harbst, K.; Staaf, J.; Lauss, M.; Karlsson, A.; Måsbäck, A.; Johansson, I.; Bendahl, P.-O.; Vallon-Christersson, J.; Törngren, T.; Ekedahl, H.; et al. Molecular profiling reveals low- and high-grade forms of primary melanoma. Clin. Cancer Res. 2012, 18, 4026–4036. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Zemek, R.M.; De Jong, E.; Chin, W.L.; Schuster, I.S.; Fear, V.S.; Casey, T.H.; Forbes, C.; Dart, S.J.; Leslie, C.; Zaitouny, A.; et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med. 2019, 11, eaav7816. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Redondo, P.; Solano, T.; Vazquez, B.; Bauza, A.; Idoate, M. Fas and Fas ligand: Expression and soluble circulating levels in cutaneous malignant melanoma. Br. J. Dermatol. 2002, 147, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Brunner, T.; Fletcher, S.M.; Green, D.R.; Ferguson, T.A. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995, 270, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Hahne, M.; Rimoldi, D.; Schröter, M.; Romero, P.; Schreier, M.; French, L.E.; Schneider, P.; Bornand, T.; Fontana, A.; Lienard, D.; et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: Implications for tumor immune escape. Science 1996, 274, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shen, Q.; Liao, H.; Fu, H.; Wang, Q.; Yu, J.; Zhang, W.; Chen, C.; Dong, Y.; Yang, X.; et al. Multi-Arm PEG/Peptidomimetic Conjugate Inhibitors of DR6/APP Interaction Block Hematogenous Tumor Cell Extravasation. Adv. Sci. 2021, 8, e2003558. [Google Scholar] [CrossRef]
- Cholewa, B.D.; Liu, X.; Ahmad, N. The role of polo-like kinase 1 in carcinogenesis: Cause or consequence? Cancer Res. 2013, 73, 6848–6855. [Google Scholar] [CrossRef]
- Su, S.; Chhabra, G.; Ndiaye, M.A.; Singh, C.K.; Ye, T.; Huang, W.; Dewey, C.N.; Setaluri, V.; Ahmad, N. PLK1 and NOTCH Positively Correlate in Melanoma and Their Combined Inhibition Results in Synergistic Modulations of Key Melanoma Pathways. Mol. Cancer Ther. 2021, 20, 161–172. [Google Scholar] [CrossRef]
- Cholewa, B.D.; Ndiaye, M.A.; Huang, W.; Liu, X.; Ahmad, N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett. 2017, 385, 179–187. [Google Scholar] [CrossRef]
- Pietraszek-Gremplewicz, K.; Simiczyjew, A.; Dratkiewicz, E.; Podgórska, M.; Styczeń, I.; Matkowski, R.; Ziętek, M.; Nowak, D. Expression level of EGFR and MET receptors regulates invasiveness of melanoma cells. J. Cell. Mol. Med. 2019, 23, 8453–8463. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Zhao, G.; Zhang, X.; Hao, M.; Hassan, S.; Zhang, M.; Zheng, H.; Yang, D.; Liu, L.; et al. IGFBP2 regulates PD-L1 expression by activating the EGFR-STAT3 signaling pathway in malignant melanoma. Cancer Lett. 2020, 477, 19–30. [Google Scholar] [CrossRef]
- Mei, Y.; Chen, M.-J.M.; Liang, H.; Ma, L. A four-gene signature predicts survival and anti-CTLA4 immunotherapeutic responses based on immune classification of melanoma. Commun. Biol. 2021, 4, 383. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yang, J.; Han, J.; He, J.; Liao, W. A novel immune checkpoint-related seven-gene signature for predicting prognosis and immunotherapy response in melanoma. Int. Immunopharmacol. 2020, 87, 106821. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhu, Z.; Lan, Y.; Duan, S.; Zhu, Z.; Zhang, X.; Li, G.; Qu, H.; Feng, Y.; Cai, H.; et al. Development and Validation of a CD8+ T Cell Infiltration-Related Signature for Melanoma Patients. Front. Immunol. 2021, 12, 659444. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Coefficient |
|---|---|
| FASLG | −0.367 |
| PLK1 | 0.287 |
| EGFR | 0.202 |
| TNFRSF21 | −0.118 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; He, J.; Chen, A.; Ran, J.; Li, J.; Chen, D.; Zhang, H. Comprehensive Analysis of Necroptosis Landscape in Skin Cutaneous Melanoma for Appealing its Implications in Prognosis Estimation and Microenvironment Status. J. Pers. Med. 2023, 13, 245. https://doi.org/10.3390/jpm13020245
Cao X, He J, Chen A, Ran J, Li J, Chen D, Zhang H. Comprehensive Analysis of Necroptosis Landscape in Skin Cutaneous Melanoma for Appealing its Implications in Prognosis Estimation and Microenvironment Status. Journal of Personalized Medicine. 2023; 13(2):245. https://doi.org/10.3390/jpm13020245
Chicago/Turabian StyleCao, Xiaoying, Jiaming He, An Chen, Jianhua Ran, Jing Li, Dilong Chen, and Hengshu Zhang. 2023. "Comprehensive Analysis of Necroptosis Landscape in Skin Cutaneous Melanoma for Appealing its Implications in Prognosis Estimation and Microenvironment Status" Journal of Personalized Medicine 13, no. 2: 245. https://doi.org/10.3390/jpm13020245
APA StyleCao, X., He, J., Chen, A., Ran, J., Li, J., Chen, D., & Zhang, H. (2023). Comprehensive Analysis of Necroptosis Landscape in Skin Cutaneous Melanoma for Appealing its Implications in Prognosis Estimation and Microenvironment Status. Journal of Personalized Medicine, 13(2), 245. https://doi.org/10.3390/jpm13020245