
Whole-Exome Sequencing Identifies Genetic Variants for Severe Adolescent Idiopathic Scoliosis in a Taiwanese Population

 , , , , , and
, , , , , and 
Abstract
1. Introduction
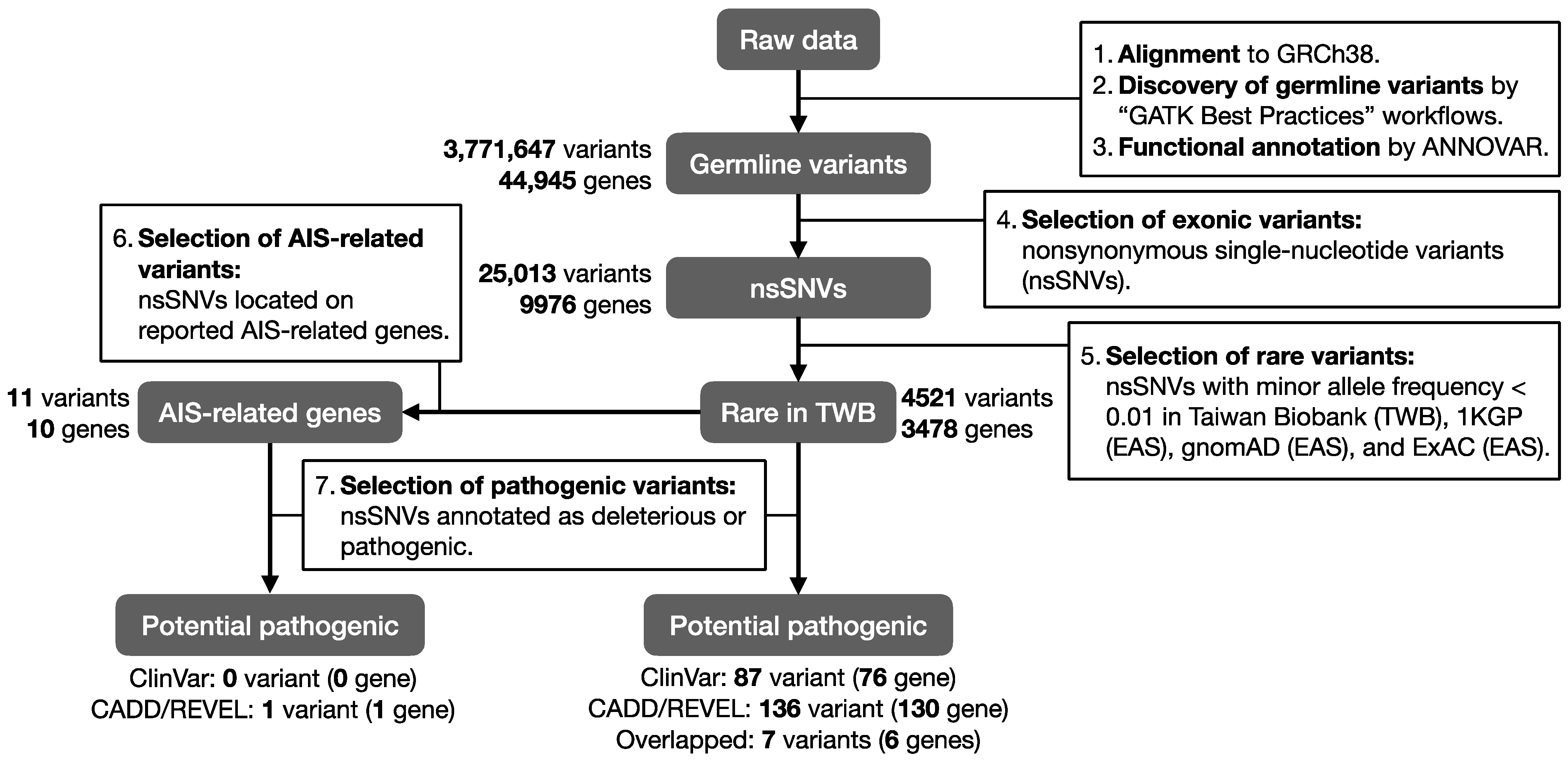
2. Materials and Methods
2.1. Study Subjects and Sample Preparation
2.2. WES and Bioinformatic Analysis
2.3. AIS Related Genes
2.4. Functional Annotation
3. Results
3.1. Patient Basal Characteristic
3.2. Pathogenic Rare Variants in Previously Reported AIS-Related Genes
3.3. Pathogenic Rare Variants on Candidate AIS Genes
3.4. Pathogenic rare Variants Carried by Patient 7
3.5. Functional Annotation of AIS Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Konieczny, M.R.; Senyurt, H.; Krauspe, R. Epidemiology of adolescent idiopathic scoliosis. J. Child Orthop. 2013, 7, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.L.; Zavala, D.C.; Ponseti, I.V. Idiopathic scoliosis: Long-term follow-up and prognosis in untreated patients. J. Bone Jt. Surg. Am. 1981, 63, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Castelein, R.M.; Chu, W.C.; Danielsson, A.J.; Dobbs, M.B.; Grivas, T.B.; Gurnett, C.A.; Luk, K.D.; Moreau, A.; Newton, P.O.; et al. Adolescent idiopathic scoliosis. Nat. Rev. Dis. Prim. 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X. Treatment of bracing for adolescent idiopathic scoliosis patients: A meta-analysis. Eur. Spine J. 2019, 28, 2012–2019. [Google Scholar] [CrossRef]
- Karavidas, N.S. Bracing for Adolescent Idiopathic Scoliosis (AIS) and Scheuermann Kyphosis: The issue of overtreatment in Greece. Scoliosis Spinal Disord. 2016, 11 (Suppl. 2), 30. [Google Scholar] [CrossRef]
- Reamy, B.V.; Slakey, J.B. Adolescent idiopathic scoliosis: Review and current concepts. Am. Fam. Physician 2001, 64, 111–116. [Google Scholar]
- Grauers, A.; Einarsdottir, E.; Gerdhem, P. Genetics and pathogenesis of idiopathic scoliosis. Scoliosis Spinal Disord. 2016, 11, 45. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kou, I.; Takahashi, A.; Johnson, T.A.; Kono, K.; Kawakami, N.; Uno, K.; Ito, M.; Minami, S.; Yanagida, H.; et al. A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 2011, 43, 1237–1240. [Google Scholar] [CrossRef]
- Kou, I.; Takahashi, Y.; Johnson, T.A.; Takahashi, A.; Guo, L.; Dai, J.; Qiu, X.; Sharma, S.; Takimoto, A.; Ogura, Y.; et al. Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet. 2013, 45, 676–679. [Google Scholar] [CrossRef]
- Ogura, Y.; Kou, I.; Miura, S.; Takahashi, A.; Xu, L.; Takeda, K.; Takahashi, Y.; Kono, K.; Kawakami, N.; Uno, K.; et al. A Functional SNP in BNC2 Is Associated with Adolescent Idiopathic Scoliosis. Am. J. Hum. Genet. 2015, 97, 337–342. [Google Scholar] [CrossRef]
- Buchan, J.G.; Alvarado, D.M.; Haller, G.E.; Cruchaga, C.; Harms, M.B.; Zhang, T.; Willing, M.C.; Grange, D.K.; Braverman, A.C.; Miller, N.H.; et al. Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum. Mol. Genet. 2014, 23, 5271–5282. [Google Scholar] [CrossRef] [PubMed]
- Haller, G.; Alvarado, D.; McCall, K.; Yang, P.; Cruchaga, C.; Harms, M.; Goate, A.; Willing, M.; Morcuende, J.A.; Baschal, E.; et al. A polygenic burden of rare variants across extracellular matrix genes among individuals with adolescent idiopathic scoliosis. Hum. Mol. Genet. 2016, 25, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Wang, L.J.; Liu, S.H.; Li, J.; Xiao, L.G.; Yang, G.T. Adiponectin regulates bone mass in AIS osteopenia via RANKL/OPG and IL6 pathway. J. Transl. Med. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Suh, K.T.; Eun, I.S.; Lee, J.S. Polymorphism in vitamin D receptor is associated with bone mineral density in patients with adolescent idiopathic scoliosis. Eur. Spine J. 2010, 19, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Chen, N.; Zhou, X.; Zhao, B.; Huang, R.; Liang, J.; Yang, X.; Chen, M.; Song, Y.; Du, Q. Alterations of the gut microbiome and plasma proteome in Chinese patients with adolescent idiopathic scoliosis. Bone 2019, 120, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Grivas, T.B.; Vasiliadis, E.; Mouzakis, V.; Mihas, C.; Koufopoulos, G. Association between adolescent idiopathic scoliosis prevalence and age at menarche in different geographic latitudes. Scoliosis 2006, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Machida, M.; Dubousset, J.; Yamada, T.; Kimura, J. Serum melatonin levels in adolescent idiopathic scoliosis prediction and prevention for curve progression–A prospective study. J. Pineal Res. 2009, 46, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhou, H.; Gao, B.; Li, Y.; Liao, Z.; Zhou, T.; Lian, C.; Wu, Z.; Su, D.; Wang, T.; et al. Estrogen promotes the onset and development of idiopathic scoliosis via disproportionate endochondral ossification of the anterior and posterior column in a bipedal rat model. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Yawn, B.P.; Yawn, R.A.; Hodge, D.; Kurland, M.; Shaughnessy, W.J.; Ilstrup, D.; Jacobsen, S.J. A population-based study of school scoliosis screening. JAMA 1999, 282, 1427–1432. [Google Scholar] [CrossRef]
- Wei, C.Y.; Yang, J.H.; Yeh, E.C.; Tsai, M.F.; Kao, H.J.; Lo, C.Z.; Chang, L.P.; Lin, W.J.; Hsieh, F.J.; Belsare, S.; et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. NPJ Genom. Med. 2021, 6, 10. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Smith, C.L.; Goldsmith, C.A.; Eppig, J.T. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005, 6, R7. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- Milla, C.P.; De Castro, C.P.; Gómez-González, C.; Martínez-Montero, P.; Pascual Pascual, S.I.; Molano Mateos, J. Myotonia congenita: Mutation spectrum of CLCN1 in Spanish patients. J. Genet. 2019, 98, 71. [Google Scholar] [CrossRef]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Udd, B.; Hackman, P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J. Neuromuscular Dis. 2016, 3, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, C.; Rowell, J.; Ferreiro, A. A rising titan: TTN review and mutation update. Hum. Mutat. 2014, 35, 1046–1059. [Google Scholar] [CrossRef]
- Rees, M.; Nikoopour, R.; Fukuzawa, A.; Kho, A.L.; Fernandez-Garcia, M.A.; Wraige, E.; Bodi, I.; Deshpande, C.; Özdemir, Ö.; Daimagüler, H.S.; et al. Making sense of missense variants in TTN-related congenital myopathies. Acta Neuropathol. 2021, 141, 431–453. [Google Scholar] [CrossRef] [PubMed]
- Pusch, M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 2002, 19, 423–434. [Google Scholar] [CrossRef]
- Reichmann, H.; Goebel, H.H.; Schneider, C.; Toyka, K.V. Familial mixed congenital myopathy with rigid spine phenotype. Muscle Nerve 1997, 20, 411–417. [Google Scholar] [CrossRef]
- Skálová, D.; Zídková, J.; Voháňka, S.; Mazanec, R.; Mušová, Z.; Vondráček, P.; Mrázová, L.; Kraus, J.; Réblová, K.; Fajkusová, L. CLCN1 mutations in Czech patients with myotonia congenita, in silico analysis of novel and known mutations in the human dimeric skeletal muscle chloride channel. PLoS ONE 2013, 8, e82549. [Google Scholar] [CrossRef]
- Rios, J.J.; Denton, K.; Yu, H.; Manickam, K.; Garner, S.; Russell, J.; Ludwig, S.; Rosenfeld, J.A.; Liu, P.; Munch, J.; et al. Saturation mutagenesis defines novel mouse models of severe spine deformity. Dis. Model. Mech. 2021, 14, dmm048901. [Google Scholar] [CrossRef]
- Stolt, C.C.; Schmitt, S.; Lommes, P.; Sock, E.; Wegner, M. Impact of transcription factor Sox8 on oligodendrocyte specification in the mouse embryonic spinal cord. Dev. Biol. 2005, 281, 309–317. [Google Scholar] [CrossRef]
- Abati, E.; Citterio, G.; Bresolin, N.; Comi, G.P.; Corti, S. Glial cells involvement in spinal muscular atrophy: Could SMA be a neuroinflammatory disease? Neurobiol. Dis. 2020, 140, 104870. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013, 136, 471–482. [Google Scholar] [CrossRef]
- Aranmolate, A.; Tse, N.; Colognato, H. Myelination is delayed during postnatal brain development in the mdx mouse model of Duchenne muscular dystrophy. BMC Neurosci. 2017, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, D.; Poulat, F.; Holinski-Feder, E.; Kooy, F.; Scherer, G. The SOX8 gene is located within 700 kb of the tip of chromosome 16p and is deleted in a patient with ATR-16 syndrome. Genomics 2000, 63, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Glaser, G.; Wernig, A.; Wegner, M.; Rosorius, O. Sox8 is a specific marker for muscle satellite cells and inhibits myogenesis. J. Biol. Chem. 2003, 278, 29769–29775. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sex | Age [Year] | Location of the Curve | Cobb Angle [Degree] |
|---|---|---|---|---|
| Pt. 1 | M | 18 | T5-T10 | 50 |
| Pt. 2 | F | 17 | T10-L3 | 47 |
| Pt. 3 | F | 16 | T2-T12 | 45 |
| Pt. 4 | F | 22 | T7-L2 | 61 |
| Pt. 5 | M | 25 | T9-L2 | 82 |
| Pt. 6 | M | 17 | T6-T12 | 72 |
| Pt. 7 | F | 19 | T12-L4/ T11-T6 | 44/45 |
| Pt. 8 | F | 24 | T12-L4 | 45 |
| Pt. 9 | F | 14 | T4-T10/T11-L4/ | 69/66 |
| Pt. 10 | F | 16 | T1-T4/T4-T12/L1-L4 | 49/86/56 |
| Pt. 11 | F | 21 | T5-T12 | 50 |
| Chr. | Position | Allele | Gene | Type | Number of Alternative Alleles in 11 AIS Patients | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | Alt. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||||
| 1 | 228097412 | G | T | ARF1 | missense | - | - | 1 | - | - | - | - | - | - | - | - |
| 3 | 65478781 | G | A | MAGI1 | missense | - | - | - | 1 | - | - | - | - | - | - | - |
| 3 | 171093913 | G | A | TNIK | missense | - | - | - | - | - | - | - | - | - | - | 1 |
| 4 | 71765499 | A | T | GC | missense | - | - | - | - | - | - | - | - | - | 1 | - |
| 5 | 128464741 | C | A | FBN2 | missense | 1 | - | - | - | - | - | - | - | - | - | - |
| 6 | 116117932 | G | A | NT5DC1 | missense | - | - | - | - | - | 1 | - | - | - | - | - |
| 7 | 22727278 | A | G | IL6 | missense | 1 | - | - | - | - | - | - | - | - | - | - |
| 8 | 3142671 | G | A | CSMD1 | missense | - | - | - | 1 | - | - | - | - | - | - | - |
| 20 | 21709343 | C | A | PAX1 | missense | - | - | - | - | - | - | - | 1 | - | - | - |
| 22 | 19761061 | C | A | TBX1 | missense | - | - | - | - | 1 | - | - | - | - | - | - |
| 22 | 19761069 | C | T | TBX1 | missense | - | 1 | - | - | - | - | - | - | - | - | - |
| Chr. | Position | Ref. | Alt. | Gene | Type | Allelic Frequency | Pathogenicity | AIS a | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1KGP | gnomAD | ExAC | TWB | ClinVar | CADD | REVEL | |||||||
| (EAS) | (EAS) | (EAS) | |||||||||||
| 5 | 156759365 | A | G | SGCD | missense | - | 0.008 | 0.0064 | 0.01 | Conflicting interpretations of pathogenicity | 20.3 | 0.865 | 5 |
| 7 | 143330810 | G | A | CLCN1 | missense | - | - | 0.0002 | - | Pathogenic/likely pathogenic | 29.5 | 0.9 | 11 |
| 8 | 104428071 | T | C | DPYS | missense | 0.003 | 0.001 | 0.0013 | 0.001 | Pathogenic | 28.1 | 0.946 | 8 |
| 8 | 104429590 | C | T | DPYS | missense | - | - | 0.0001 | - | Likely pathogenic | 35 | 0.894 | 9 |
| 11 | 47346297 | C | T | MYBPC3 | missense | 0.004 | 0.0026 | 0.0045 | 0.001 | Conflicting interpretations of pathogenicity | 24.9 | 0.757 | 9 |
| 11 | 77189442 | G | C | MYO7A | missense | 0.004 | 0.0057 | 0.0042 | 0.0085 | Conflicting interpretations of pathogenicity | 28.5 | 0.853 | 9 |
| 20 | 13816520 | T | G | NDUFAF5 | missense | - | 0.001 | 0.0006 | 0.0015 | Conflicting interpretations of pathogenicity | 27.7 | 0.887 | 3 |
| Pathway Category | No. of Reference Genes | No. of Overlap with AIS Candidate Genes | p Value | q Value (FDR) |
|---|---|---|---|---|
| Hypertrophic cardiomyopathy | 83 | 9 | 3.61 × 10−6 | 1.18 × 10−3 |
| Arrhythmogenic right ventricular cardiomyopathy | 72 | 8 | 1.07 × 10−5 | 1.75 × 10−3 |
| Dilated cardiomyopathy | 90 | 8 | 5.55 × 10−5 | 6.03 × 10−3 |
| Lysosome | 123 | 8 | 4.90 × 10−4 | 3.99 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.-R.; Chou, P.-H.; Huang, K.-J.; Ting, J.; Liu, C.-Y.; Chou, W.-H.; Lin, G.-H.; Chang, J.-G.; Ikegawa, S.; Wang, S.-T.; et al. Whole-Exome Sequencing Identifies Genetic Variants for Severe Adolescent Idiopathic Scoliosis in a Taiwanese Population. J. Pers. Med. 2023, 13, 32. https://doi.org/10.3390/jpm13010032
Lin M-R, Chou P-H, Huang K-J, Ting J, Liu C-Y, Chou W-H, Lin G-H, Chang J-G, Ikegawa S, Wang S-T, et al. Whole-Exome Sequencing Identifies Genetic Variants for Severe Adolescent Idiopathic Scoliosis in a Taiwanese Population. Journal of Personalized Medicine. 2023; 13(1):32. https://doi.org/10.3390/jpm13010032
Chicago/Turabian StyleLin, Min-Rou, Po-Hsin Chou, Kuei-Jung Huang, Jafit Ting, Chia-Ying Liu, Wan-Hsuan Chou, Gan-Hong Lin, Jan-Gowth Chang, Shiro Ikegawa, Shih-Tien Wang, and et al. 2023. "Whole-Exome Sequencing Identifies Genetic Variants for Severe Adolescent Idiopathic Scoliosis in a Taiwanese Population" Journal of Personalized Medicine 13, no. 1: 32. https://doi.org/10.3390/jpm13010032
APA StyleLin, M.-R., Chou, P.-H., Huang, K.-J., Ting, J., Liu, C.-Y., Chou, W.-H., Lin, G.-H., Chang, J.-G., Ikegawa, S., Wang, S.-T., & Chang, W.-C. (2023). Whole-Exome Sequencing Identifies Genetic Variants for Severe Adolescent Idiopathic Scoliosis in a Taiwanese Population. Journal of Personalized Medicine, 13(1), 32. https://doi.org/10.3390/jpm13010032