Abstract
The process of tooth formation is a series of reciprocal interactions between the ectoderm and mesoderm, and it is believed that many genetic factors are involved in this complex process. More than a dozen genes have been identified in non-syndromic tooth agenesis; however, the genetic etiology underlying tooth agenesis is not fully understood yet. In this study, we identified two novel LRP6 mutations in two non-syndromic oligodontia families. Both probands had 16 and 17 missing teeth in their permanent dentition. Mutational analysis identified a de novo frameshift mutation by a 1-bp insertion in exon 9 (NM_002336.2: c.1870dupA, p.(Met624Asnfs*29)) and a splicing donor site mutation in intron 8 (c.1762+2T>C). An in vitro splicing assay confirmed the deletion of exon 8, and the deletion would result in a frameshift. Due to the premature termination codons introduced by the frameshift, both mutant transcripts would be degraded by nonsense-mediated mRNA decay, resulting in haploinsufficiency.
1. Introduction
Tooth formation is a result of a series of reciprocal ectodermal mesenchymal interactions [1]. Genetic defects or environmental assaults can cause tooth agenesis (TA) or a malformed tooth, such as peg lateralis, cone-shaped tooth, or taurodontism [2]. Due to the shared developmental origin, failure of tooth formation sometimes accompanies alterations in other ectodermally derived structures, such as the nails, hair, and sweat glands [3]. Therefore, TA can be classified as syndromic or non-syndromic TA, but the defects in the same gene can cause both TAs without a clear-cut boundary [4].
Oligodontia is a term that indicates a rare genetic condition in which six or more teeth (excluding wisdom teeth) are congenitally missing [5]. Anodontia, in which all teeth are missing, is extremely rare, but hypodontia, in which five or less teeth are missing, is relatively common. The pattern of TA, the locations of the conserved and missing teeth, could suggest candidate disease-causing genes [6]. Genetic heterogeneity and variable expressivity, however, make it hard to predict an exact culprit that causes selective tooth agenesis [7].
Even though the molecular pathology underlying TA is complex and is not fully understood yet, there are three major signaling pathways involved in TA: WNT/β-catenin, EDA/NF-κB, and TGF-β/BMP [8]. Aberrant signaling caused by mutant ligands, receptors, and any other components that relay the signal has been shown to cause TA, and the list includes MSX1 (OMIM *142983, msh homeobox 1) [9], PAX9 (OMIM *167416, paired box 9) [10], AXIN2 (OMIM *604025, axin 2) [11], EDA (OMIM *300451, ectodysplasin A) [12], EDAR (OMIM *604095, ectodysplasin A receptor) [13], EDARADD (OMIM *606603, EDAR associated death domain) [14], WNT10A (OMIM *606268, Wnt family member 10A) [15], WNT10B (OMIM *601906, Wnt family member 10B) [16], GREM2 (OMIM *608832, gremlin 2, DAN family BMP antagonist) [17], BMP4 (OMIM *112262, bone morphogenetic protein 4) [18], SMOC2 (OMIM *607223, SPARC-related modular calcium binding 2) [19], LRP6 (OMIM *603507, LDL receptor-related protein 6) [20], and KREMEN1 (OMIM *609898, kringle containing transmembrane protein 1) [21].
In this study, we recruited families with oligodontia and performed mutational analysis by whole-exome sequencing. The mutational analyses revealed two novel LRP6 mutations, and an in vitro splicing assay verified the effect of the mutation. This report expands the mutational spectrum of the LRP6 gene. A literature review with the identified mutations compared the effect of the LRP6 mutations to advance our understanding of the molecular pathogenesis of TA.
2. Materials and Methods
2.1. Human Subject Enrollment
Protocols of the study and patient consent were reviewed and approved by the institutional review board of Seoul National University Dental Hospital (CRI05003G and 9 December 2021). Informed consent was obtained from all participating individuals after explaining the nature of the study. The pedigree was drawn with the family histories taken. Clinical examination and sample collection were performed according to the principles in the Declaration of Helsinki.
2.2. Genomic DNA Isolation and Whole-Exome Sequencing
Genomic DNA was isolated from 2 mL of peripheral blood by a conventional method with the NucleoSpin Blood L kit (Macherey-Nagel GmbH & Co., Düren, Germany) according to the manufacturer’s instructions. After measuring the quality and quantity using the NanoDrop1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA), the DNA samples of the probands were submitted for whole-exome sequencing (Yale Center for Mendelian Genomics, West Haven, CT, USA, and Theragen Etex Bio Institute, Suwon-si, Gyeonggi-do, Korea). After exome capturing, paired-end sequencing reads were generated.
2.3. Bioinformatic Analysis
Obtained sequencing reads (Table 1) were processed using a series of bioinformatic analyses as previously described [22]. Briefly, after trimming to remove the adapter sequences, the reads were aligned to the reference human genome assembly (hg38). Bioinformatics analysis programs, such as Samtools and Genome Analysis Tool Kit, were used to get a list of sequence variants [23,24]. The dbSNP build 147 database was used for the annotation of the sequence variants, and the annotated variants were filtered with a minor allele frequency of 0.01.

Table 1.
Statistics for exome sequencing.
2.4. Sanger Sequencing
The identified mutations and the segregation among the family members were confirmed by Sanger sequencing. The following primer pairs for the LRP6 gene were used: sense (5′-GTCCTTCTGTGCCCCTTTTA-3′) and antisense (5′-TCTCCCTTTTAGTCCCTAGCTTT-3′) primers for exon 9 and sense (5′-TCATCATGTAATTTTGAGAAAGCA-3′) and antisense (5′-TCTCCCTTTTAGTCCCTAGCTTT-3′) primers for exon 8. Sanger sequencing was performed for all participating family members at Macrogen (Seoul, Korea). The LRP6 mutations were submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 26 July 2022), Accession ID: SCV002549922 and SCV002549923).
2.5. Human Identification and Paternity Test
DNA samples of the trio of family 1 (the proband and parents) were submitted for human identification (Macrogen). A short tandem repeat multiplex assay was performed with the AmpFISTR®Identifiler® KIT (Applied Biosystems, Foster City, CA, USA) using 15 tetranucleotide repeat loci and the Amelogenin gender-determining marker: all thirteen of the required loci for the Combined DNA Index System and two additional loci (D2S1338 and D19S433). Genotyping data were obtained with the Applied Biosystems® 3730/3730xl DNA Analyzer and analyzed with GeneMapper ID v3.2 (Applied Biosystems).
2.6. In Vitro Splicing Assay
The wild-type and mutant genomic fragments of the LRP6 gene were amplified from a DNA sample of the proband of family 2 using the Pfu Plus 5x PCR Master mix (Elpis Biotech, Daejeon, Korea) and cloned into the TOPcloner Blunt V2 vector (Enzynomics, Seoul, Korea) using the same primers as in the exon 8 sequencing (including exons 8 and 9). After the sequences were confirmed, the wild-type and mutant fragments were subcloned into the pSPL3 splicing vector after double digestion with NotI and BamHI restriction endonucleases. COS-7 cells were transiently transfected with the wild-type and mutant pSPL3 vectors, and total RNA was isolated after 36 hours, and the cDNA was synthesized. RT-PCR (sense 5′-TCTGAGTCACCTGGACAACC-3′ and antisense 5′-AGGAAAGCCTCTGGGACAAT-3′) was performed with the EzPCR™ Basic 5x Master Mix (Elpis Biotech). Amplification bands were excised from an agarose gel following electrophoresis, purified, and characterized by direct DNA sequencing.
2.7. Dimensional In Silico Modeling
To better understand the molecular characteristics of the LRP6 protein, three-dimensional modeling was performed with the PyMOL Molecular Graphics System (Version 1.8.2.3, Schrödinger, LLC., New York, NY, USA). Structure modules (3s94, 3s8z, 5gje, and 6l6r) were downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (https://www.rcsb.org/ (accessed on 26 July 2022)).
3. Results
3.1. Clinical Phenotype and Mutational Analysis of Family 1
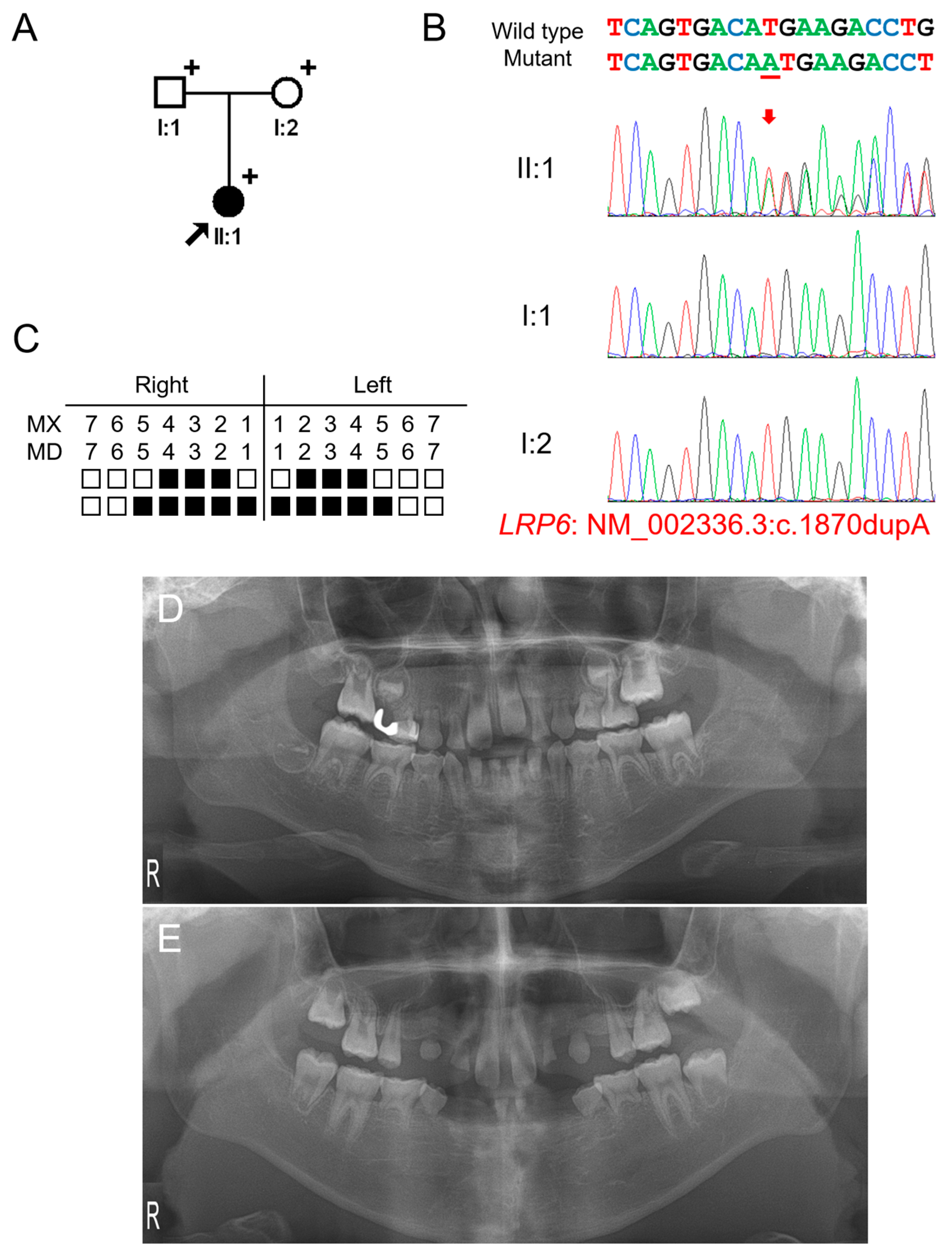
The proband of family 1 was a 5-year-11-month-old female from a non-consanguineous Korean family (Figure 1). Pregnancy and delivery were uneventful, and she had no remarkable past medical history. Her deciduous dentition was normal without malformed or missing teeth; therefore, her family did not know about the missing multiple teeth in her permanent dentition. They were told about it when an examination was performed at a local dental clinic. The right maxillary second deciduous molar was exfoliated, and the distal-shoe space maintainer was placed. The left maxillary first molar exhibited an ectopic eruption with the resorption of the distal root of the left second deciduous molar. The maxillary first molars had taurodontism. She had 16 permanent teeth missing excluding the third molars (which were also missing), but no other syndromic features including oral exostosis. Her parents were normal without any missing teeth. Therefore, a recessive or de novo dominant mutation was suspected.
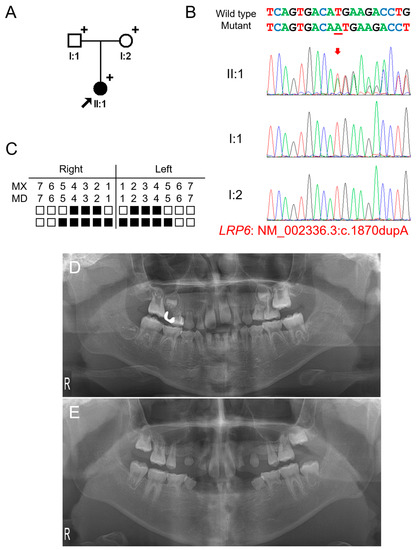
Figure 1.
Pedigree, chromatograms, and panoramic radiographs of family 1. (A) Pedigree of family 1. Black symbols indicate affected individuals, and the proband is indicated by a black arrow. Plus signs above the symbols indicate participating individuals. (B) Sequencing chromatograms of the participating individuals of family 1. Wild-type and mutant nucleotide sequences are shown above the chromatograms. Nucleotide affected by the mutation is underlined. The location of the mutation is indicated with a red arrow. Individual identifications are indicated on the left side of each chromatogram. (C) Summary chart of the missing teeth of the proband. Black box indicates a missing tooth. Tooth number is shown above the boxes (MX: maxilla, MN: mandible). (D) Panoramic radiograph of the proband at age 5 years 11 months shows multiple missing teeth. Maxillary left first molar exhibits ectopic eruption. The maxillary first molars have taurodontism. (E) Panoramic radiograph of the proband at age 12 years 5 months.
Mutational analysis of the whole-exome data resulted in three variants in genes related to TA: heterozygous WNT10A (NM_025216: c.637G>A, p.(Gly213Ser)), homozygous EDAR (NM_022336: c.1138A>C, p.(Ser380Arg)) and heterozygous LRP6 (NM_002336.2: c.1870dupA, p.(Met624Asnfs*29)) variants. The WNT10A variant was inherited from the healthy mother and previously reported to cause autosomal recessive oligodontia [25]. Therefore, this heterozygous variant is not considered as a disease-causing mutation. The parents were heterozygous for the EDAR variant, and the proband was homozygous. This variant was previously reported as a disease-causing mutation to cause autosomal dominant inheritance with high conservation and in silico prediction values. However, it has been shown this variant exhibits an increased EDAR signaling output to a similar level of the variant p.(Val370Ala). Therefore, this variant also is not considered as a disease-causing mutation, even though it is homogeneous. The heterozygous LRP6 variant was a 1-bp insertion in exon 9 of 23 exons and predicted to cause a change in the reading frame (p.(Met624Asnfs*29)). This frameshift would result in a premature termination codon (PTC) in the early exon; therefore, it would be degraded by the nonsense-mediated mRNA decay system (NMDS) [26], resulting in the haploinsufficiency of LRP6. This mutation was not found in the parents, and the paternity test confirmed that this mutation occurred spontaneously. Therefore, this frameshift mutation caused by a de novo 1-bp nucleotide insertion is believed to cause severe oligodontia in the proband.
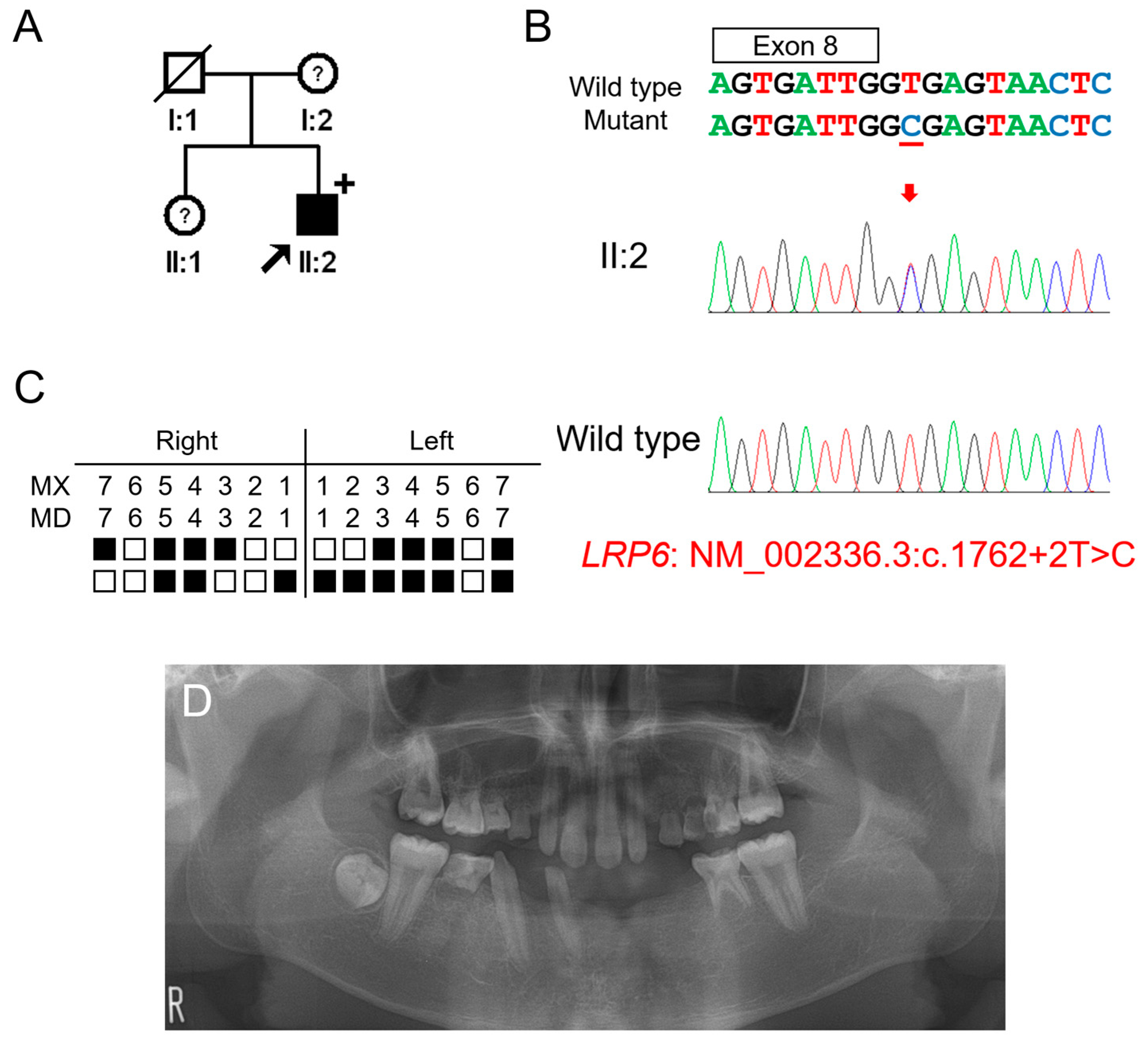
3.2. Clinical Phenotype and Mutational Analysis of Family 2
The proband of family 2 was a 14-year-5-month-old male from a non-consanguineous Korean family (Figure 2). He had no other remarkable past medical history. His mother and sister were reported to have missing teeth; however, examination and confirmation were not available. He had 17 missing permanent teeth excluding his missing third molars. Maxillary lateral incisors were peg lateralis bilaterally, and the first molars showed the characteristic feature of taurodontism. Whether the deciduous teeth were missing could not be confirmed. There were no other syndromic features including oral exostosis.
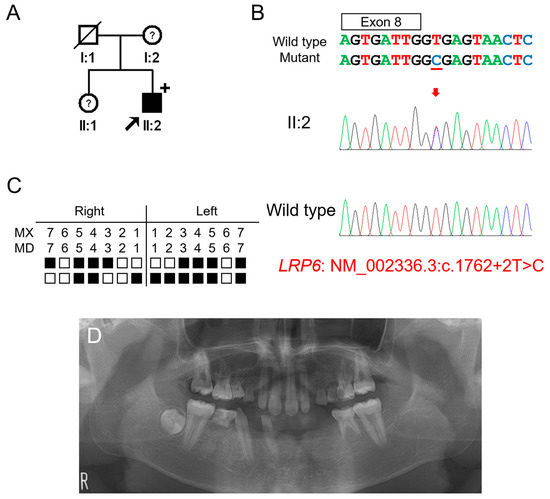
Figure 2.
Pedigree, chromatograms and panoramic radiographs of family 2. (A) Pedigree of family 2. Black symbols indicate affected individuals, and the proband is indicated by a black arrow. Plus sign above the symbol indicates participating individuals. Symbols with a question mark inside indicate individuals whose phenotypes are not confirmed. (B) Sequencing chromatograms of the proband of family 2. Wild-type and mutant nucleotide sequences are shown above the chromatograms. Exon is indicated by a box above the nucleotide sequences. Nucleotide affected by the mutation is underlined. The location of the mutation is indicated with a red arrow. Individual identifications are indicated on the left side of each chromatogram. (C) Summary chart of the missing teeth of the proband. Black box indicates a missing tooth. Tooth number is shown above the boxes (MX: maxilla, MN: mandible). (D) Panoramic radiograph of the proband at age 14 years 5 months shows multiple missing teeth. First molars exhibit taurodontism.
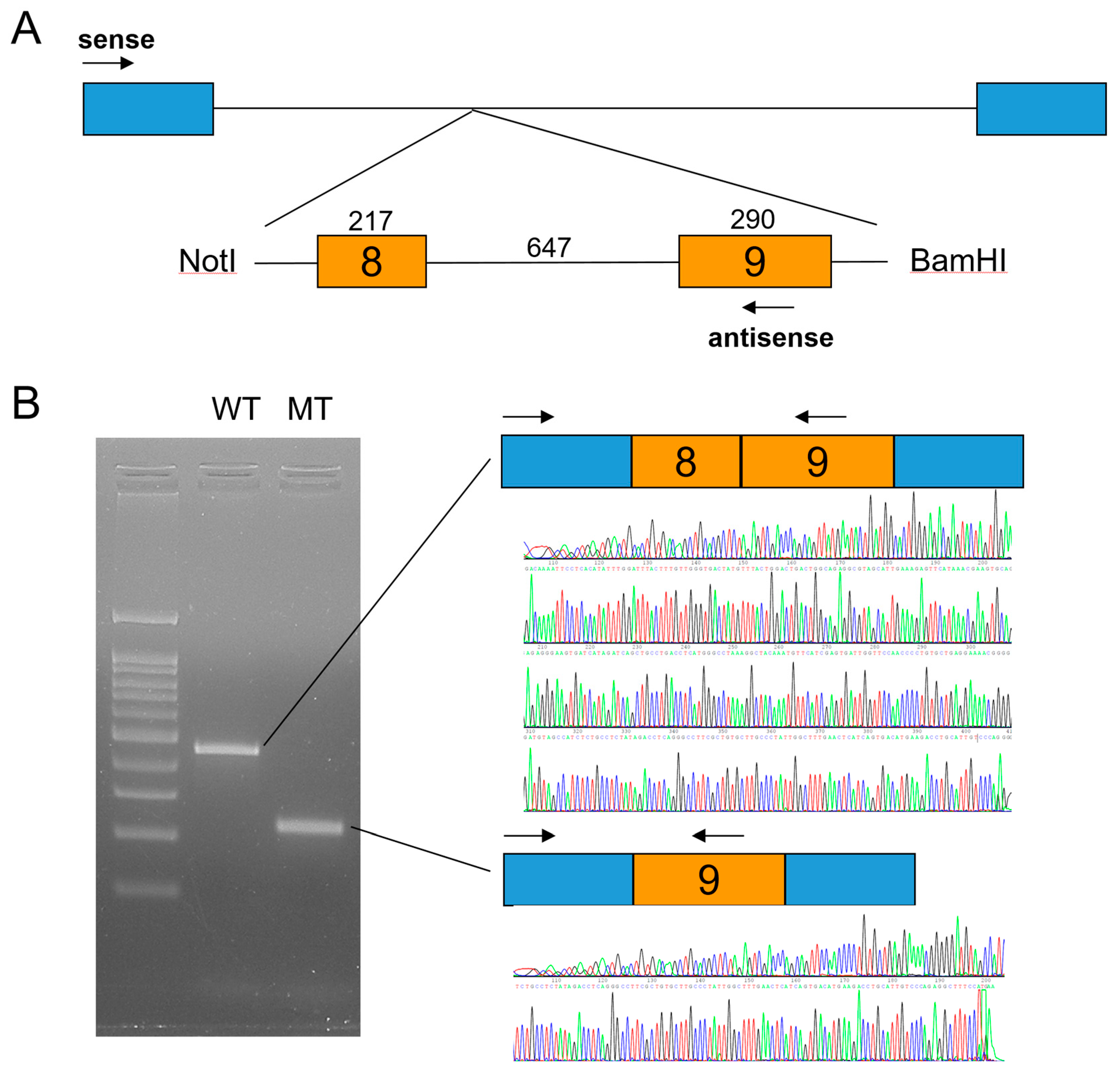
Candidate gene sequencing of the MSX1, WNT10A, and EDA genes was performed but failed to identify any disease-causing mutations. Whole-exome sequencing revealed no other variants in the genes causing TA, except for an LRP6 variant, a transitional change of thymine to cytosine at the canonical splicing donor site sequence (GT to GC) in intron 8 (NM_002336.2: c.1762+2T>C). An in vitro splicing assay confirmed the deletion of exon 8 during pre-mRNA splicing (Figure 3). Deletion of the 217-bp exon 8 would cause a change in the reading frame and introduce a PTC in exon 9. This mutant mRNA transcript would be degraded by the NMDS, resulting in the haploinsufficiency of LRP6 as in the case of family 1.
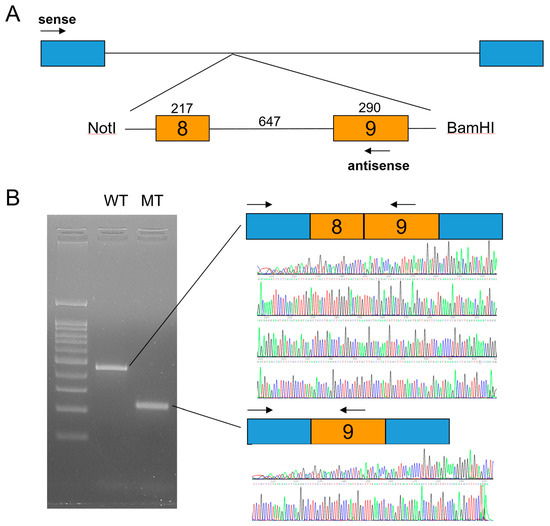
Figure 3.
In vitro splicing assay. (A) Minigene cloning strategy. A genomic fragment including exons 8 and 9 of LRP6 was subcloned into the pSPL3 vector with double digestion using NotI and BamHI restriction endonucleases. Boxes indicate exons, and horizontal lines indicate introns. The number of the exon is in the box, and the length of the exon and intron is shown above the boxes and line. Locations of the primer binding site are indicated with arrows (sense and antisense). (B) Agarose gel image of the splicing assay of the wild type (WT) and mutant (MT). Left lane is the DNA ladder. Wild-type and mutant names are shown above the gel image. Wild-type vector resulted in a normal splicing product including exons 8 and 9. Mutation (c.1762+2T>C) resulted in an exon 8 deletion. Sequencing chromatograms of the alternative splicing band are shown below.
4. Discussion
In this study, two novel LRP6 mutations were identified, and the mutational effect was confirmed by an in vitro splicing assay. Both mutations would result in the haploinsufficiency caused by mutant mRNA degradation by the NMDS. Both probands exhibited severe TA (16 and 17 missing) in their permanent dentition. Usually, the TA phenotype caused by LRP6 mutations does not, minimal if any (lateral incisors), exhibit any missing deciduous teeth [27]. Even though missing deciduous teeth could not be confirmed in the proband of family 2, the proband of family 1 does not have any missing deciduous teeth. A previous study reported taurodontism in about one-third of affected individuals [20], and the probands in our study also showed taurodontism: mandibular second molars in the proband of family 1 and all four first permanent molars in the proband of family 2. LRP6 mutations related to tooth agenesis is listed in Table 2.
Table 2.
LRP6 mutations related to tooth agenesis.
Expressivity is variable, especially wide in some missense mutations [33,38]; furthermore, an individual with incomplete penetrance was reported in a familial case with a frameshift mutation (c.1144_1145dupAG), which is supposed to be degraded by the NMDS [20]. A recent study demonstrated that there could be many more unknown genetic players involved in TA because mutations were identified in only about 25% of the study participants (12 out of 49 subjects) [36]. Additionally, a possible synergistic effect of the LRP6 and WNT10A mutations has been also suggested [30]; therefore, it would be possible that multigenic effects among genes, even unknown and yet to be identified, are involved in TA.
Without proper functional studies, interpretation of an identified potential candidate variant sometimes misleads us to misunderstand the molecular pathogenesis of TA (including other complex disease entities), especially if the variant is rare and the amino acid encoded is highly conserved among species, because in silico predictions would favor the pathologic effect if so [42,43]. Due to the limitation of the repertoire of the known genes, it is possible that it might not be disease-causing but just very rare, and the disease is caused by other unknown gene(s).
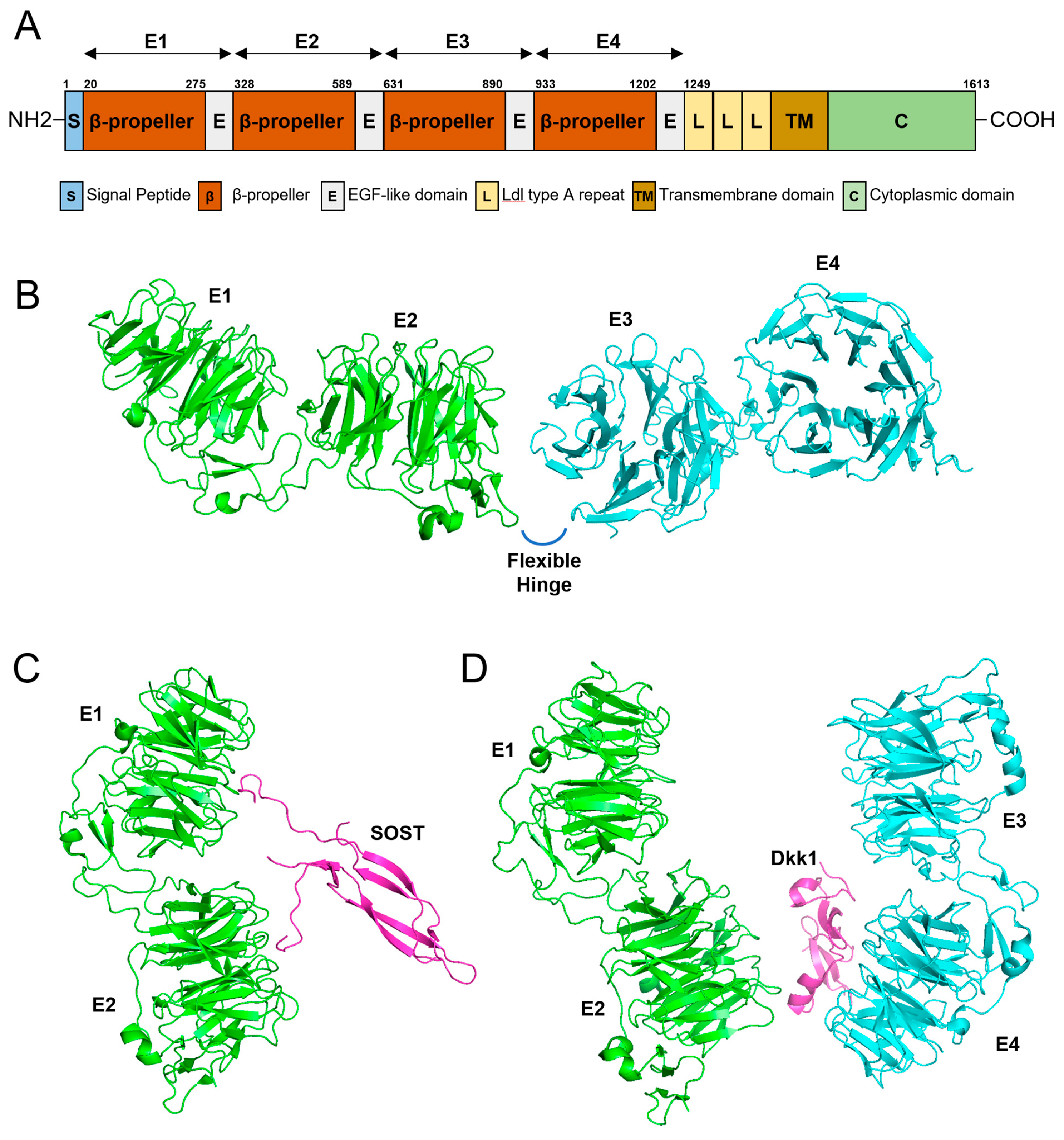
The domain structure of the LRP6 protein shows interesting features (Figure 4). LRP6 is a single-pass transmembrane protein with an N-terminal ectodomain with four characteristic β-propeller/EGF-like domain repeats (E1, E2, E3, and E4) and a C-terminus cytoplasmic domain [20]. There is a hinge-like flexibility between the E1E2 and E3E4 structures, and this flexibility enables conformational freedom if there is no ligand bound [44]. This free movement hampers the unwanted formation of homo or heterobinding of LRP6 to form an uncontrolled signaling platform [45]. With a ligand bound such as Wnt, restriction to free movement and conformational change enables the formation of oligomerization to convey controlled signals [46]. Interestingly, a conformational change caused by Dkk1 binding prevents oligomerization [44].
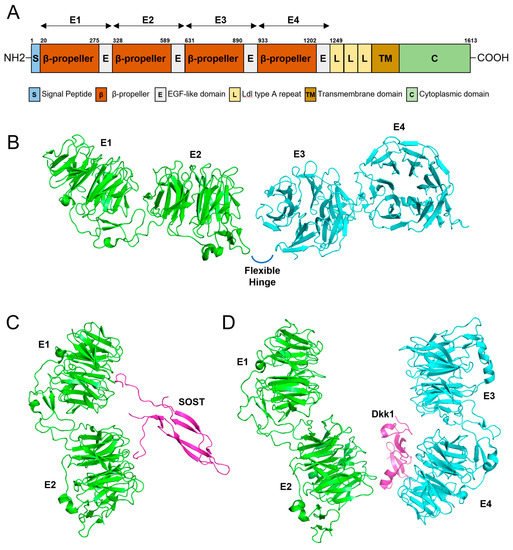
Figure 4.
Gene diagram and 3D modeling. (A) Domain structure of LRP6. Amino acid numbers are shown above the diagram. Four β-propeller/EGF-like domain repeats (E1, E2, E3, and E4) are shown above the diagram with arrows. (B) 3D modeling of the four β-propeller/EGF-like domain repeats (E1, E2, E3, and E4). There is a flexible hinge between E2 and E3. (C) Three-dimensional modeling of E1 and E2 binding to SOST. (D) Three-dimensional modeling of the ectodomain of LRP6 complexed with Dkk1.
LPR6 mutations sometimes accompany syndromic features such as sparse hair like in ectodermal dysplasia, a cleft lip and palate, hand preaxial polydactyly, and an orofacial cleft (OFC) (Table 2) [27,29,31,32,35,38]. The mutation in OFC is a deletion causing a frameshift in the last exon; however, due to the PTC location in the last exon, the mutant transcript would escape from the NMDS and produce a truncated protein with novel amino acids in the cytoplasmic C-terminal region [27]. This would be more harmful than the other frameshift mutations degraded by the NMDS by a dominant negative effect. Ligand binding would be normal with the intact ectodomain, but intracellular signaling would be disturbed or trigger a malfunction. Some mutations show interesting features, such as a high bone mass (with or without torus palatinus) with minimal TA (maxillary only or all four lateral incisors missing) [31,32]. The location of the mutations is in the E1 domain and may suggest involvement of certain factors to cause the high bone mass. Likewise, expanding the mutational spectrum of LRP6 and related phenotypes may provide insight into the complex regulation and function of LRP6-mediated signaling pathways.
5. Conclusions
In this study, we recruited families with non-syndromic oligodontia and identified two novel LRP6 mutations: a de novo frameshift mutation by a 1-bp insertion in exon 9 (c.1870dupA, p.(Met624Asnfs*29)) and a splicing donor site mutation in intron 8 (c.1762+2T>C). A minigene splicing assay confirmed that the effect of the splicing mutation and the mRNAs of both mutations are predicted to be degraded by the NMDS due to PTC. Further studies including genetic and functional studies are warranted to understand and characterize the complex mechanism of tooth formation.
Author Contributions
Y.L., W.C. and Y.J.K. contributed to analysis and interpretation, data acquisition, and critically revised the manuscript; J.-W.K. contributed to the conception, design, data acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean government (MEST) (NRF-2018R1A5A2024418 and NRF-2020R1A2C2100543).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of the Seoul National University Dental Hospital (CRI05003G and 9 December 2021).
Informed Consent Statement
Informed consent was obtained from all individual participants included in the study.
Data Availability Statement
The data presented in this study are openly available in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar (accessed on 26 July 2022)), Accession ID: SCV002549922 and SCV002549923.
Acknowledgments
We are grateful to all family members who participated in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thesleff, I. Epithelial-mesenchymal signalling regulating tooth morphogenesis. J. Cell Sci. 2003, 116, 1647–1648. [Google Scholar] [CrossRef] [PubMed]
- Al-Ani, A.H.; Antoun, J.S.; Thomson, W.M.; Merriman, T.R.; Farella, M. Hypodontia: An Update on Its Etiology, Classification, and Clinical Management. BioMed Res. Int. 2017, 2017, 9378325. [Google Scholar] [CrossRef] [PubMed]
- García-Martín, P.; Hernández-Martín, A.; Torrelo, A. Ectodermal dysplasias: A clinical and molecular review. Actas Dermo-Sifiliogr. 2013, 104, 451–470. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. Part A 2019, 179, 442–447. [Google Scholar] [CrossRef] [PubMed]
- De Coster, P.J.; Marks, L.A.; Martens, L.C.; Huysseune, A. Dental agenesis: Genetic and clinical perspectives. J. Oral Pathol. Med. 2009, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.P.; Bruneau, M.H.; Toupenay, S.; Kerner, S.; Berdal, A.; Cormier-Daire, V.; Hadj-Rabia, S.; Coudert, A.E.; de La Dure-Molla, M. Patterns of Dental Agenesis Highlight the Nature of the Causative Mutated Genes. J. Dent. Res. 2018, 97, 1306–1316. [Google Scholar] [CrossRef]
- Williams, M.A.; Letra, A. The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis. Genes 2018, 9, 255. [Google Scholar] [CrossRef]
- Yu, M.; Wong, S.-W.; Han, D.; Cai, T. Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Dis. 2019, 25, 646–651. [Google Scholar] [CrossRef]
- Vastardis, H.; Karimbux, N.; Guthua, S.W.; Seidman, J.G.; Seidman, C.E. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat. Genet. 1996, 13, 417–421. [Google Scholar] [CrossRef]
- Stockton, D.W.; Das, P.; Goldenberg, M.; D’Souza, R.N.; Patel, P.I. Mutation of PAX9 is associated with oligodontia. Nat. Genet. 2000, 24, 18–19. [Google Scholar] [CrossRef]
- Lammi, L.; Arte, S.; Somer, M.; Järvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 Cause Familial Tooth Agenesis and Predispose to Colorectal Cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Monreal, A.W.; Ferguson, B.M.; Headon, D.J.; Street, S.L.; Overbeek, P.A.; Zonana, J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat. Genet. 1999, 22, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef]
- Adaimy, L.; Chouery, E.; Mégarbané, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Mégarbané, A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef]
- Yu, P.; Yang, W.; Han, D.; Wang, X.; Guo, S.; Li, J.; Li, F.; Zhang, X.; Wong, S.-W.; Bai, B.; et al. Mutations in WNT10B Are Identified in Individuals with Oligodontia. Am. J. Hum. Genet. 2016, 99, 195–201. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Kaewgahya, M.; Hatsadaloi, A.; Vogel, P.; Kawasaki, K.; Ohazama, A.; Ketudat Cairns, J.R. GREMLIN 2 Mutations and Dental Anomalies. J. Dent. Res. 2015, 94, 1646–1652. [Google Scholar] [CrossRef]
- Huang, Y.; Lu, Y.; Mues, G.; Wang, S.; Bonds, J.; D’Souza, R. Functional evaluation of a novel tooth agenesis-associated bone morphogenetic protein 4 prodomain mutation. Eur. J. Oral Sci. 2013, 121, 313–318. [Google Scholar] [CrossRef]
- Bloch-Zupan, A.; Jamet, X.; Etard, C.; Laugel, V.; Muller, J.; Geoffroy, V.; Strauss, J.-P.; Pelletier, V.; Marion, V.; Poch, O.; et al. Homozygosity Mapping and Candidate Prioritization Identify Mutations, Missed by Whole-Exome Sequencing, in SMOC2, Causing Major Dental Developmental Defects. Am. J. Hum. Genet. 2011, 89, 773–781. [Google Scholar] [CrossRef]
- Massink, M.P.; Créton, M.A.; Spanevello, F.; Fennis, W.M.; Cune, M.S.; Savelberg, S.M.; Nijman, I.J.; Maurice, M.M.; van den Boogaard, M.-J.H.; van Haaften, G. Loss-of-Function Mutations in the WNT Co-receptor LRP6 Cause Autosomal-Dominant Oligodontia. Am. J. Hum. Genet. 2015, 97, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Issa, Y.A.; Kamal, L.; Rayyan, A.A.; Dweik, D.; Pierce, S.; Lee, M.K.; King, M.C.; Walsh, T.; Kanaan, M. Mutation of KREMEN1, a modulator of Wnt signaling, is responsible for ectodermal dysplasia including oligodontia in Palestinian families. Eur. J. Hum. Genet. 2016, 24, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Lee, Y.; Zhang, H.; Seymen, F.; Koruyucu, M.; Bayrak, S.; Tuloglu, N.; Simmer, J.P.; Hu, J.C.; Kim, J.W. Translated Mutant DSPP mRNA Expression Level Impacts the Severity of Dentin Defects. J. Pers. Med. 2022, 12, 1002. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Park, H.; Song, J.-S.; Shin, T.J.; Hyun, H.-K.; Kim, Y.-J.; Kim, J.-W. WNT10A mutations causing oligodontia. Arch. Oral Biol. 2019, 103, 8–11. [Google Scholar] [CrossRef]
- Brogna, S.; Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 107–113. [Google Scholar] [CrossRef]
- Ockeloen, C.W.; Khandelwal, K.D.; Dreesen, K.; Ludwig, K.U.; Sullivan, R.; Van Rooij, I.A.L.M.; Thonissen, M.; Swinnen, S.; Phan, M.; Conte, F.; et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet. Med. 2016, 18, 1158–1162. [Google Scholar] [CrossRef]
- Kantaputra, P.; Jatooratthawichot, P.; Chintakanon, K.; Intachai, W.; Pradermdutsadeeporn, P.; Adisornkanj, P.; Tongsima, S.; Ngamphiw, C.; Olsen, B.; Tucker, A.S.; et al. Mutations in LRP6 highlight the role of WNT signaling in oral exostoses and dental anomalies. Arch. Oral Biol. 2022, 142, 105514. [Google Scholar] [CrossRef]
- Yu, M.; Fan, Z.; Wong, S.W.; Sun, K.; Zhang, L.; Liu, H.; Feng, H.; Liu, Y.; Han, D. LRP6 Dynamic Expression in Tooth Development and Mutations in Oligodontia. J. Dent. Res. 2021, 100, 415–422. [Google Scholar] [CrossRef]
- Chu, K.-Y.; Wang, Y.-L.; Chou, Y.-R.; Chen, J.-T.; Wang, Y.-P.; Simmer, J.P.; Hu, J.C.-C.; Wang, S.-K. Synergistic Mutations of LRP6 and WNT10A in Familial Tooth Agenesis. J. Pers. Med. 2021, 11, 1217. [Google Scholar] [CrossRef]
- Whyte, M.P.; McAlister, W.H.; Zhang, F.; Bijanki, V.N.; Nenninger, A.; Gottesman, G.S.; Lin, E.L.; Huskey, M.; Duan, S.; Dahir, K.; et al. New explanation for autosomal dominant high bone mass: Mutation of low-density lipoprotein receptor-related protein 6. Bone 2019, 127, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Brance, M.L.; Brun, L.R.; Cóccaro, N.M.; Aravena, A.; Duan, S.; Mumm, S.; Whyte, M.P. High bone mass from mutation of low-density lipoprotein receptor-related protein 6 (LRP6). Bone 2020, 141, 115550. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Zheng, Y.; Zhao, X.; Lin, S.; Zhang, Q.; Zhang, X. A novel missense mutation of LRP6 identified by whole-exome sequencing in a Chinese family with non-syndromic tooth agenesis. Orthod. Craniofac. Res. 2020, 24, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, H.; Camhi, H.; Seymen, F.; Koruyucu, M.; Kasimoglu, Y.; Kim, J.W.; Kim-Berman, H.; Yuson, N.M.R.; Benke, P.J.; et al. Analyses of oligodontia phenotypes and genetic etiologies. Int. J. Oral Sci. 2021, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, M.; Sun, K.; Fan, Z.; Liu, H.; Feng, H.; Liu, Y.; Han, D. Rare phenotype: Hand preaxial polydactyly associated with LRP6-related tooth agenesis in humans. npj Genom. Med. 2021, 6, 93. [Google Scholar] [CrossRef]
- Keskin, G.; Karaer, K.; Uçar Gündoğar, Z. Targeted next-generation sequencing (NGS) analysis of mutations in nonsyndromic tooth agenesis candidate genes: Analysis of a Turkish cohort. J. Orofac. Orthop. 2021. [Google Scholar] [CrossRef]
- Goto, H.; Kimura, M.; Machida, J.; Ota, A.; Nakashima, M.; Tsuchida, N.; Adachi, J.; Aoki, Y.; Tatematsu, T.; Takahashi, K.; et al. A novel LRP6 variant in a Japanese family with oligodontia. Hum. Genome Var. 2021, 8, 30. [Google Scholar] [CrossRef]
- Huang, Y.X.; Gao, C.Y.; Zheng, C.Y.; Chen, X.; Yan, Y.S.; Sun, Y.Q.; Dong, X.Y.; Yang, K.; Zhang, D.L. Investigation of a Novel LRP6 Variant Causing Autosomal-Dominant Tooth Agenesis. Front Genet. 2021, 12, 688241. [Google Scholar] [CrossRef]
- Basha, M.; Demeer, B.; Revencu, N.; Helaers, R.; Theys, S.; Saba, S.B.; Boute, O.; Devauchelle, B.; Francois, G.; Bayet, B.; et al. Whole exome sequencing identifies mutations in 10% of patients with familial non-syndromic cleft lip and/or palate in genes mutated in well-known syndromes. J. Med. Genet. 2018, 55, 449–458. [Google Scholar] [CrossRef]
- Dinckan, N.; Du, R.; Petty, L.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Paine, I.; Baugh, E.H.; Erdem, A.P.; Kayserili, H.; Doddapaneni, H.; et al. Whole-Exome Sequencing Identifies Novel Variants for Tooth Agenesis. J. Dent. Res. 2018, 97, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.; Fennis, W.; De Leeuw, N.; Cune, M.; Willemze, A.; Rosenberg, A.; Ploos van Amstel, H.-K.; Créton, M.; van den Boogaard, M.-J. Concurrent manifestation of oligodontia and thrombocytopenia caused by a contiguous gene deletion in 12p13.2: A three-generation clinical report. Mol. Genet. Genom. Med. 2019, 7, e679. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K.; Mihara, E.; Tamura-Kawakami, K.; Miyazaki, N.; Maeda, S.; Hirai, H.; Thompson, S.; Iwasaki, K.; Takagi, J. Conformational Freedom of the LRP6 Ectodomain Is Regulated by N-glycosylation and the Binding of the Wnt Antagonist Dkk1. Cell Rep. 2017, 18, 32–40. [Google Scholar] [CrossRef]
- Ahn, V.E.; Chu, M.L.; Choi, H.J.; Tran, D.; Abo, A.; Weis, W.I. Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev. Cell 2011, 21, 862–873. [Google Scholar] [CrossRef]
- Chen, S.; Bubeck, D.; MacDonald, B.T.; Liang, W.X.; Mao, J.H.; Malinauskas, T.; Llorca, O.; Aricescu, A.R.; Siebold, C.; He, X.; et al. Structural and functional studies of LRP6 ectodomain reveal a platform for Wnt signaling. Dev. Cell 2011, 21, 848–861. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).