
Natural History and Molecular Characteristics of Korean Patients with Mucopolysaccharidosis Type III

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. MPS III Subtype Classification: Enzyme Assays and Genetic Tests
2.2. Statistical Analysis
2.3. Ethics Statement
3. Results
3.1. Clinical Presentation
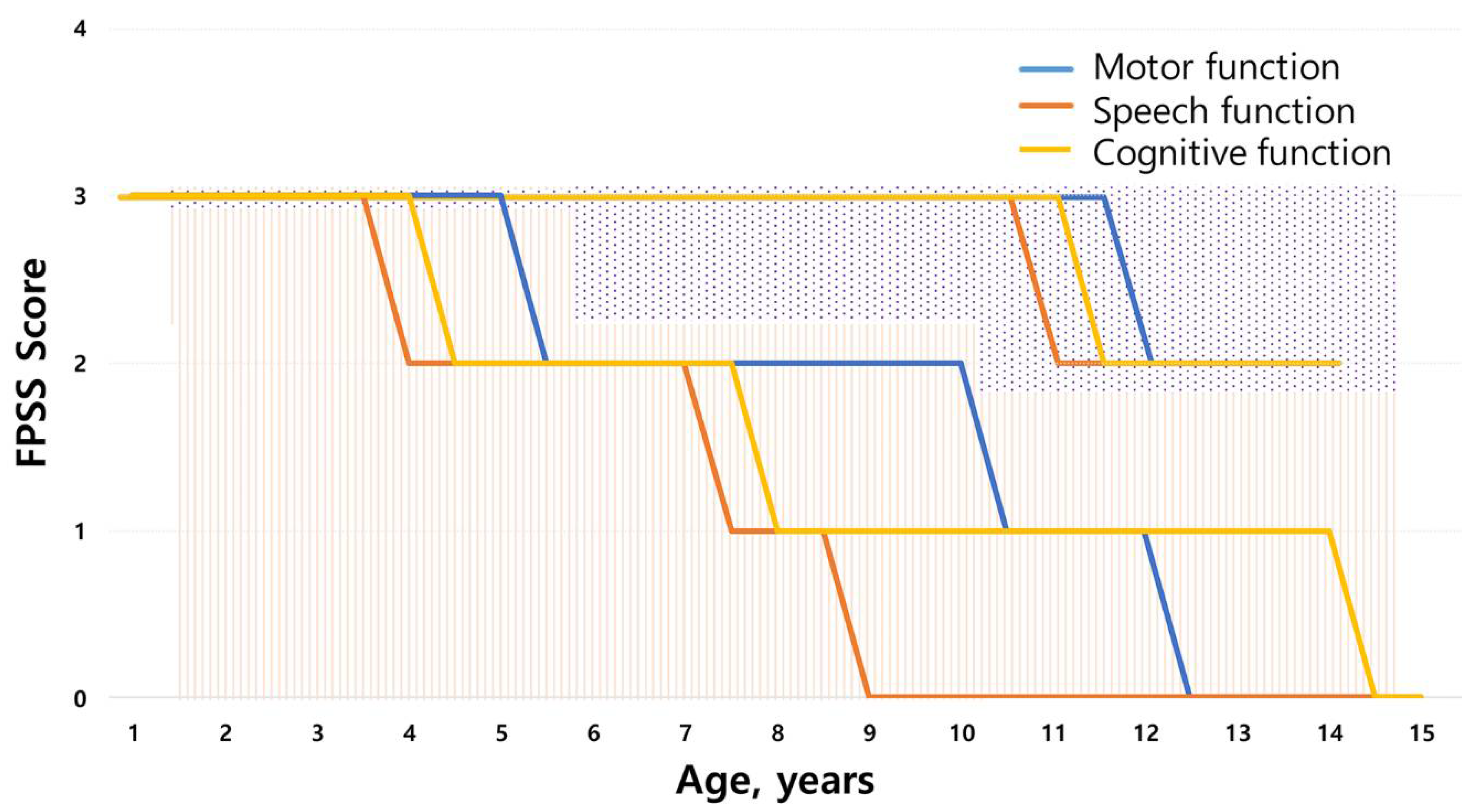
3.2. Neurodegenerative Symptoms
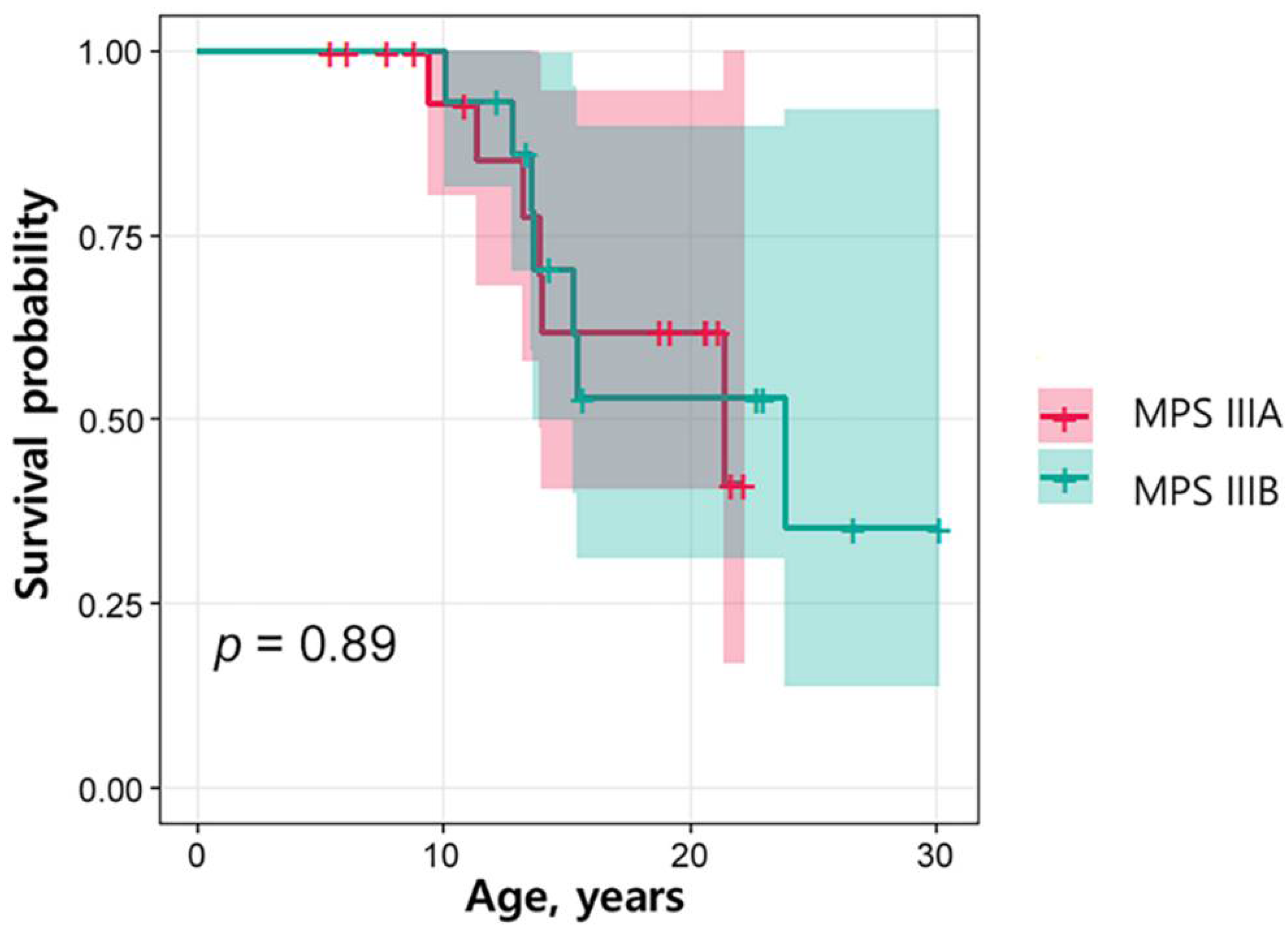
3.3. Survival Analysis by Subtype
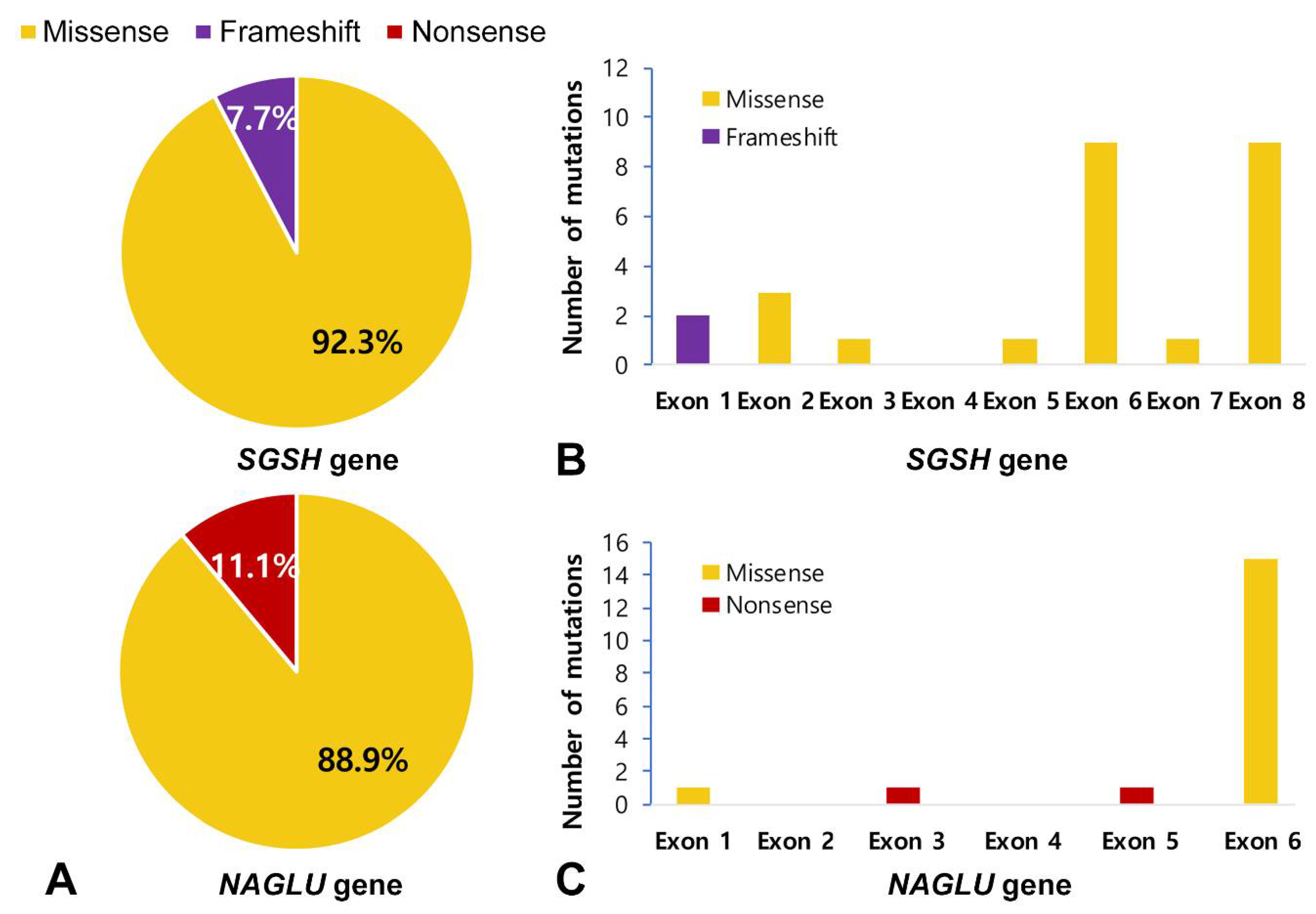
3.4. Genetic Analysis
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
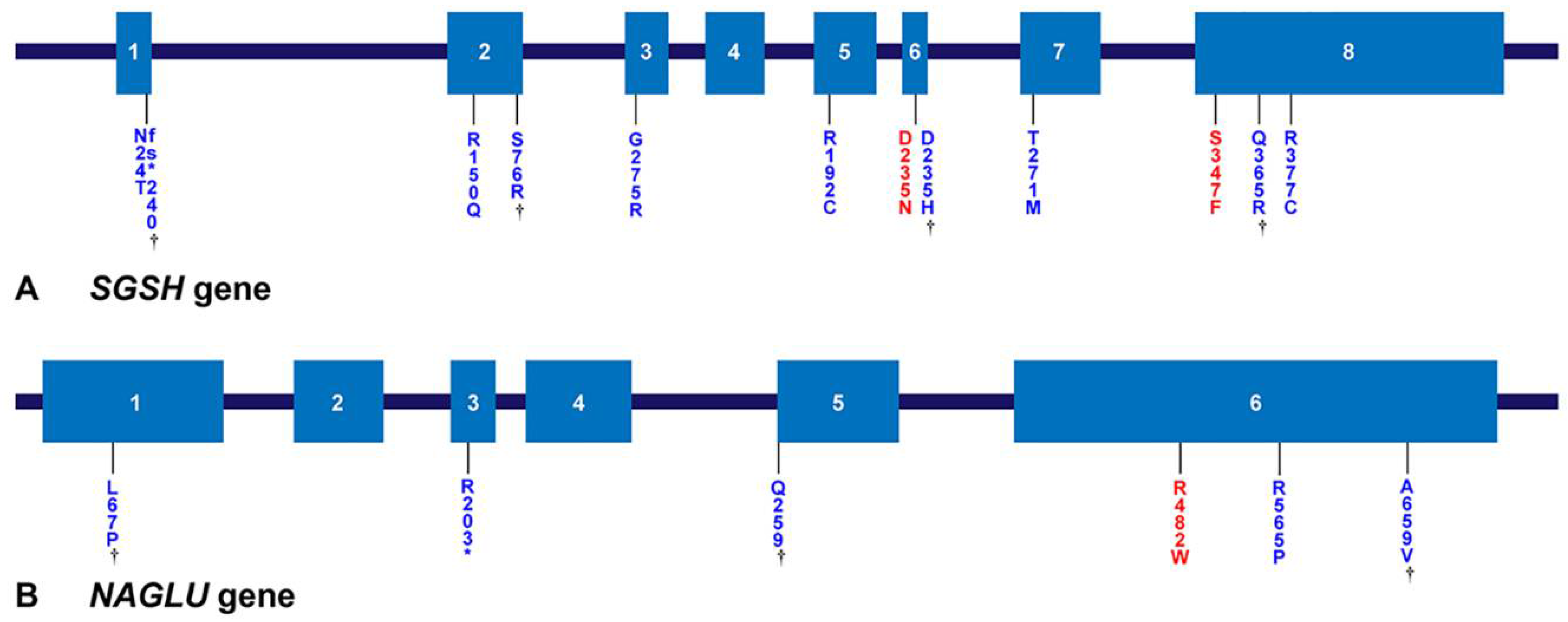{kind=link}
| Gene | Reference Sequences | DNA Nucleotide Change | Predicted Protein Change | Common [Reference] | Patient Number |
|---|---|---|---|---|---|
| SGSH | NM_000199.5 NP_000190.1 | c.1129C > T | p.Arg377Cys | Di Natale et al. [37] | 9A |
| c.1094A > G | p.Gln365Arg | Novel | 9A | ||
| c.1040C > T | p.Ser347Phe | Miyazaki et al. [38] | 1A, 2A 3A, 4A, 7A, 10A £ | ||
| c.823G > A | p.Gly275Arg | Heron et al. [8] | 15A | ||
| c.812C > T | p.Thr271Met | Heron et al. [8] | 5A | ||
| c.703G > A | p.Asp235Asn | Beesley et al. [41] | 1A, 4A, 5A, 12A, 14A, 17A | ||
| c.703G > C | p.Asp235His | Novel | 3A, 7A, 12A | ||
| c.544C > T | p.Arg192Cys | Di Natale et al. [37] | 2A | ||
| c.449G > A | p.Arg150Gln | Bunge et al. [35] | 15A, 17A | ||
| c.228C > G | p.Ser76Arg | Novel | 14A | ||
| c.69delG | p.Asn24Thrfs*240 | Novel | 8A£ | ||
| NAGLU | NM_000263.4 NP_000254.2 | c.1976C > T | p.Ala659Val | Novel | 1B |
| c.1694G > C | p.Arg565Pro | Weber et al. [42] | 12B, 14B £ | ||
| c.1444C > T | p.Arg482Trp | Bunge et al. [43] | 1B, 2B £, 5B £, 6B, 8B, 11B £, 13B £ | ||
| c.775C > T | p.Gln259* | Novel | 12B | ||
| c.607C > T | p.Arg203* | Schmidtchen et al. [44] | 8B | ||
| c.200T > C | p.Leu67Pro | Novel | 6B | ||
| HGSNAT | NM_152419.3 NP_689632.2 | c.234+1G > A | IVS2+1G > A | Canals et al. [45] | 1C, 2C |
| c.1150C > T | p.Arg384* | Ruijter et al. [23] | 1C, 2C |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shapiro, E.G.; Nestrasil, I.; Delaney, K.A.; Rudser, K.; Kovac, V.; Nair, N.; Richard, C.W., III; Haslett, P.; Whitley, C.B. A Prospective Natural History Study of Mucopolysaccharidosis Type IIIA. J. Pediatr. 2016, 170, 278–287.e4. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F. The mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Huh, H.J.; Seo, J.Y.; Cho, S.Y.; Ki, C.S.; Lee, S.Y.; Kim, J.W.; Park, H.D.; Jin, D.K. The first Korean case of mucopolysaccharidosis IIIC (Sanfilippo syndrome type C) confirmed by biochemical and molecular investigation. Ann. Lab. Med. 2013, 33, 75–79. [Google Scholar] [CrossRef]
- Emre, S.; Terzioglu, M.; Tokatli, A.; Coskun, T.; Ozalp, I.; Weber, B.; Hopwood, J.J. Sanfilippo syndrome in Turkey: Identification of novel mutations in subtypes A and B. Hum. Mutat. 2002, 19, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymanska, A.; Czartoryska, B.; Gorska, D.; Piesiewicz-Grzonkowska, E. Type III D mucopolysaccharidosis (Sanfilippo D): Clinical course and symptoms. Acta Paediatr. Jpn. 1998, 40, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Zelei, T.; Csetneki, K.; Voko, Z.; Siffel, C. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J. Rare Dis. 2018, 13, 53. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Ruijter, G.J.; van Diggelen, O.P.; Poorthuis, B.J.; Wijburg, F.A. Sanfilippo syndrome: A mini-review. J. Inherit Metab. Dis. 2008, 31, 240–252. [Google Scholar] [CrossRef]
- Heron, B.; Mikaeloff, Y.; Froissart, R.; Caridade, G.; Maire, I.; Caillaud, C.; Levade, T.; Chabrol, B.; Feillet, F.; Ogier, H.; et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am. J. Med. Genet. A 2011, 155a, 58–68. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chuang, C.K.; Lee, C.L.; Tu, R.Y.; Lo, Y.T.; Chiu, P.C.; Niu, D.M.; Fang, Y.Y.; Chen, T.L.; Tsai, F.J.; et al. Mucopolysaccharidosis III in Taiwan: Natural history, clinical and molecular characteristics of 28 patients diagnosed during a 21-year period. Am. J. Med. Genet. A 2018, 176, 1799–1809. [Google Scholar] [CrossRef]
- Kong, W.; Meng, Y.; Zou, L.; Yang, G.; Wang, J.; Shi, X. Mucopolysaccharidosis III in Mainland China: Natural history, clinical and molecular characteristics of 34 patients. J. Pediatr. Endocrinol. Metab. 2020, 33, 793–802. [Google Scholar] [CrossRef]
- Mitchell, J.; Berger, K.I.; Borgo, A.; Braunlin, E.A.; Burton, B.K.; Ghotme, K.A.; Kircher, S.G.; Molter, D.; Orchard, P.J.; Palmer, J.; et al. Unique medical issues in adult patients with mucopolysaccharidoses. Eur. J. Intern. Med. 2016, 34, 2–10. [Google Scholar] [CrossRef]
- Buhrman, D.; Thakkar, K.; Poe, M.; Escolar, M.L. Natural history of Sanfilippo syndrome type A. J. Inherit Metab. Dis. 2014, 37, 431–437. [Google Scholar] [CrossRef] [PubMed]
- van de Kamp, J.J.; Niermeijer, M.F.; von Figura, K.; Giesberts, M.A. Genetic heterogeneity and clinical variability in the Sanfilippo syndrome (types A, B, and C). Clin. Genet. 1981, 20, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Fedele, A.O. Sanfilippo syndrome: Causes, consequences, and treatments. Appl. Clin. Genet. 2015, 8, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Breen, C.; Heap, F.; Rust, S.; de Ruijter, J.; Tump, E.; Marchal, J.P.; Pan, L.; Qiu, Y.; Chung, J.K.; et al. A phase 1/2 study of intrathecal heparan-N-sulfatase in patients with mucopolysaccharidosis IIIA. Mol. Genet. Metab. 2016, 118, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zerah, M.; Gougeon, M.L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: An uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017, 16, 712–720. [Google Scholar] [CrossRef]
- Delgadillo, V.; O’Callaghan Mdel, M.; Gort, L.; Coll, M.J.; Pineda, M. Natural history of Sanfilippo syndrome in Spain. Orphanet J. Rare Dis. 2013, 8, 189. [Google Scholar] [CrossRef]
- Meyer, A.; Kossow, K.; Gal, A.; Muhlhausen, C.; Ullrich, K.; Braulke, T.; Muschol, N. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics 2007, 120, e1255–e1261. [Google Scholar] [CrossRef]
- Martins, C.; de Medeiros, P.F.V.; Leistner-Segal, S.; Dridi, L.; Elcioglu, N.; Wood, J.; Behnam, M.; Noyan, B.; Lacerda, L.; Geraghty, M.T.; et al. Molecular characterization of a large group of Mucopolysaccharidosis type IIIC patients reveals the evolutionary history of the disease. Hum. Mutat. 2019, 40, 1084–1100. [Google Scholar] [CrossRef]
- CDC’s Developmental Milestones. Available online: https://www.cdc.gov/ncbddd/actearly/milestones/index.html (accessed on 15 January 2022).
- World Health Organization. Motor Development Milestones. Available online: https://www.who.int/tools/child-growth-standards/standards/motor-development-milestones (accessed on 15 January 2022).
- Shapiro, E.G.; Eisengart, J.B. The natural history of neurocognition in MPS disorders: A review. Mol. Genet. Metab. 2021, 133, 8–34. [Google Scholar] [CrossRef]
- Ruijter, G.J.; Valstar, M.J.; van de Kamp, J.M.; van der Helm, R.M.; Durand, S.; van Diggelen, O.P.; Wevers, R.A.; Poorthuis, B.J.; Pshezhetsky, A.V.; Wijburg, F.A. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol. Genet. Metab. 2008, 93, 104–111. [Google Scholar] [CrossRef]
- Hult, M.; Darin, N.; von Dobeln, U.; Mansson, J.E. Epidemiology of lysosomal storage diseases in Sweden. Acta Paediatr. 2014, 103, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Malm, G.; Mansson, J.E. Mucopolysaccharidosis type III (Sanfilippo disease) in Sweden: Clinical presentation of 22 children diagnosed during a 30-year period. Acta Paediatr. 2010, 99, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Lee, C.L.; Chang, C.Y.; Chiu, P.C.; Chien, Y.H.; Niu, D.M.; Tsai, F.J.; Hwu, W.L.; Lin, S.J.; Lin, J.L.; et al. Survival and diagnostic age of 175 Taiwanese patients with mucopolysaccharidoses (1985–2019). Orphanet J. Rare Dis. 2020, 15, 314. [Google Scholar] [CrossRef]
- Kuiper, G.A.; Meijer, O.L.M.; Langereis, E.J.; Wijburg, F.A. Failure to shorten the diagnostic delay in two ultra-orphan diseases (mucopolysaccharidosis types I and III): Potential causes and implications. Orphanet J. Rare Dis. 2018, 13, 2. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Wegrzyn, G.; Burton, B.K.; Tylki-Szymanska, A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013, 102, 462–470. [Google Scholar] [CrossRef]
- Wolfenden, C.; Wittkowski, A.; Jones, S.A.; Rust, S.; Hare, D.J. Autism Spectrum Disorder symptomatology in children with Mucopolysaccharide Disease Type III. Br. J. Learn. Disabil. 2018, 47, 5–11. [Google Scholar] [CrossRef]
- Shapiro, E.; King, K.; Ahmed, A.; Rudser, K.; Rumsey, R.; Yund, B.; Delaney, K.; Nestrasil, I.; Whitley, C.; Potegal, M. The Neurobehavioral Phenotype in Mucopolysaccharidosis Type IIIB: An Exploratory Study. Mol. Genet. Metab. Rep. 2016, 6, 41–47. [Google Scholar] [CrossRef]
- Global Variome Shared LOVD. SGSH Gene. Available online: https://www.LOVD.nl/SGSH (accessed on 15 January 2022).
- Global Variome Shared LOVD. NAGLU Gene. Available online: https://www.LOVD.nl/NAGLU (accessed on 15 January 2022).
- Weber, B.; van de Kamp, J.J.; Kleijer, W.J.; Guo, X.H.; Blanch, L.; van Diggelen, O.P.; Wevers, R.; Poorthuis, B.J.; Hopwood, J.J. Identification of a common mutation (R245H) in Sanfilippo A patients from The Netherlands. J. Inherit Metab. Dis. 1998, 21, 416–422. [Google Scholar] [CrossRef]
- Bunge, S.; Ince, H.; Steglich, C.; Kleijer, W.J.; Beck, M.; Zaremba, J.; van Diggelen, O.P.; Weber, B.; Hopwood, J.J.; Gal, A. Identification of 16 sulfamidase gene mutations including the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo A). Hum. Mutat. 1997, 10, 479–485. [Google Scholar] [CrossRef]
- Montfort, M.; Vilageliu, L.; Garcia-Giralt, N.; Guidi, S.; Coll, M.J.; Chabás, A.; Grinberg, D. Mutation 1091delC is highly prevalent in Spanish Sanfilippo syndrome type A patients. Hum. Mutat. 1998, 12, 274–279. [Google Scholar] [CrossRef]
- Di Natale, P.; Balzano, N.; Esposito, S.; Villani, G.R. Identification of molecular defects in Italian Sanfilippo A patients including 13 novel mutations. Hum. Mutat. 1998, 11, 313–320. [Google Scholar] [CrossRef]
- Miyazaki, T.; Masuda, N.; Waragai, M.; Motoyoshi, Y.; Kurokawa, K.; Yuasa, T. An adult Japanese Sanfilippo A patient with novel compound heterozygous S347F and D444G mutations in the sulphamidase gene. J. Neurol. Neurosurg. Psychiatry 2002, 73, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Guo, X.H.; Wraith, J.E.; Cooper, A.; Kleijer, W.J.; Bunge, S.; Hopwood, J.J. Novel mutations in Sanfilippo A syndrome: Implications for enzyme function. Hum. Mol. Genet. 1997, 6, 1573–1579. [Google Scholar] [CrossRef]
- Lee-Chen, G.J.; Lin, S.P.; Ko, M.H.; Chuang, C.K.; Chen, C.P.; Lee, H.H.; Cheng, S.C.; Shen, C.H.; Tseng, K.L.; Li, C.L. Identification and characterization of mutations underlying Sanfilippo syndrome type A (mucopolysaccharidosis type IIIA). Clin. Genet. 2002, 61, 192–197. [Google Scholar] [CrossRef]
- Beesley, C.E.; Young, E.P.; Vellodi, A.; Winchester, B.G. Mutational analysis of Sanfilippo syndrome type A (MPS IIIA): Identification of 13 novel mutations. J. Med. Genet. 2000, 37, 704–707. [Google Scholar] [CrossRef]
- Weber, B.; Guo, X.H.; Kleijer, W.J.; van de Kamp, J.J.; Poorthuis, B.J.; Hopwood, J.J. Sanfilippo type B syndrome (mucopolysaccharidosis III B): Allelic heterogeneity corresponds to the wide spectrum of clinical phenotypes. Eur. J. Hum. Genet. 1999, 7, 34–44. [Google Scholar] [CrossRef]
- Bunge, S.; Knigge, A.; Steglich, C.; Kleijer, W.J.; van Diggelen, O.P.; Beck, M.; Gal, A. Mucopolysaccharidosis type IIIB (Sanfilippo B): Identification of 18 novel alpha-N-acetylglucosaminidase gene mutations. J. Med. Genet. 1999, 36, 28–31. [Google Scholar]
- Schmidtchen, A.; Greenberg, D.; Zhao, H.G.; Li, H.H.; Huang, Y.; Tieu, P.; Zhao, H.Z.; Cheng, S.; Zhao, Z.; Whitley, C.B.; et al. NAGLU mutations underlying Sanfilippo syndrome type B. Am. J. Hum. Genet. 1998, 62, 64–69. [Google Scholar] [CrossRef][Green Version]
- Canals, I.; Elalaoui, S.C.; Pineda, M.; Delgadillo, V.; Szlago, M.; Jaouad, I.C.; Sefiani, A.; Chabás, A.; Coll, M.J.; Grinberg, D.; et al. Molecular analysis of Sanfilippo syndrome type C in Spain: Seven novel HGSNAT mutations and characterization of the mutant alleles. Clin. Genet. 2011, 80, 367–374. [Google Scholar] [CrossRef]
- Yogalingam, G.; Hopwood, J.J. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: Diagnostic, clinical, and biological implications. Hum. Mutat. 2001, 18, 264–281. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Kimura, M.; Lan, H.T.; Takaura, N.; Yamano, T. Molecular analysis of the alpha-N-acetylglucosaminidase gene in seven Japanese patients from six unrelated families with mucopolysaccharidosis IIIB (Sanfilippo type B), including two novel mutations. J. Hum. Genet. 2002, 47, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Chinen, Y.; Tohma, T.; Izumikawa, Y.; Uehara, H.; Ohta, T. Sanfilippo type B syndrome: Five patients with an R565P homozygous mutation in the alpha-N-acetylglucosaminidase gene from the Okinawa islands in Japan. J. Hum. Genet. 2005, 50, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.G.; Aronovich, E.L.; Whitley, C.B. Genotype-phenotype correspondence in Sanfilippo syndrome type B. Am. J. Hum. Genet. 1998, 62, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Feldhammer, M.; Durand, S.; Mrazova, L.; Boucher, R.M.; Laframboise, R.; Steinfeld, R.; Wraith, J.E.; Michelakakis, H.; van Diggelen, O.P.; Hrebicek, M.; et al. Sanfilippo syndrome type C: Mutation spectrum in the heparan sulfate acetyl-CoA: Alpha-glucosaminide N-acetyltransferase (HGSNAT) gene. Hum. Mutat. 2009, 30, 918–925. [Google Scholar] [CrossRef]
- Hrebicek, M.; Mrazova, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Noskova, L.; Hartmannova, H.; Ivanek, R.; Cizkova, A.; Poupetova, H.; et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef]
- de Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]





| Characteristic | Total (N = 34) | IIIA (N = 18) | IIIB (N = 14) | p-Value | IIIC (N = 2) |
|---|---|---|---|---|---|
| Sex | >0.99 | ||||
| Male | 20 (58.9%) | 11 (61.1%) | 8 (57.1%) | 1 (50%) | |
| Female | 14 (41.1%) | 7 (38.9%) | 6 (42.9%) | 1 (50%) | |
| Follow-up period (mean years ± SD) | 12.6 ± 6.1 | 12.3 ± 5.8 | 13.6 ± 6.5 | 0.58 | 11.2 ± 0.6 |
| Age at symptom onset (mean years ± SD) | 2.8 ± 0.8 | 3.1 ± 0.9 | 2.6 ± 0.7 | 0.10 | 2.0 ± 0.2 |
| Age at diagnosis (mean years ± SD) | 6.3 ± 2.2 | 6.3 ± 2.3 | 6.5 ± 2.0 | 0.77 | 3.7 ± 1.4 |
| Diagnosis lag time (mean years ± SD) | 3.6 ± 2.5 | 3.5 ± 2.9 | 3.9 ± 1.9 | 0.29 | 1.7 ± 1.2 |
| Cardiac evaluation by TTE (positive finding/test available, %) | NA | ||||
| MR | 15/19 (78.9%) | 10/14 (71.4%) | 5/5 (100.0%) | 0.53 | |
| TR | 16/19 (84.2%) | 11/14 (78.6%) | 5/5 (100.0%) | 0.53 | |
| MV thickening | 13/19 (68.4%) | 9/14 (64.3%) | 4/5 (80.0%) | >0.99 | |
| Abdominal US (positive finding / test available, %) | |||||
| Hepatosplenomegaly | 17/21 (80.9%) | 9/12 (75%) | 6/7 (85.7%) | >0.99 | 2/2 (100%) |
| SMA syndrome | 6/21 (28.6%) | 4/12 (33.3%) | 2/7 (28.6%) | 0.51 | 0 |
| Clinical findings | |||||
| Age at first walking without assistance, median years (IQR Q1,Q3) | 1.3 (1.23, 1.6) | 1.25 (1, 1.37) | 1.55 (1.42, 1.6) | 0.002 * | 1.3, 1.0 |
| Age of first seizure (mean years ± SD) | 10.0 ± 2.9 | 9.8 ± 2.8 | 10.3 ± 3.2 | 0.739 | NA |
| History of AEDs medication | 26 (76.5%) | 15 (83.3%) | 11 (78.6%) | >0.99 | 0 |
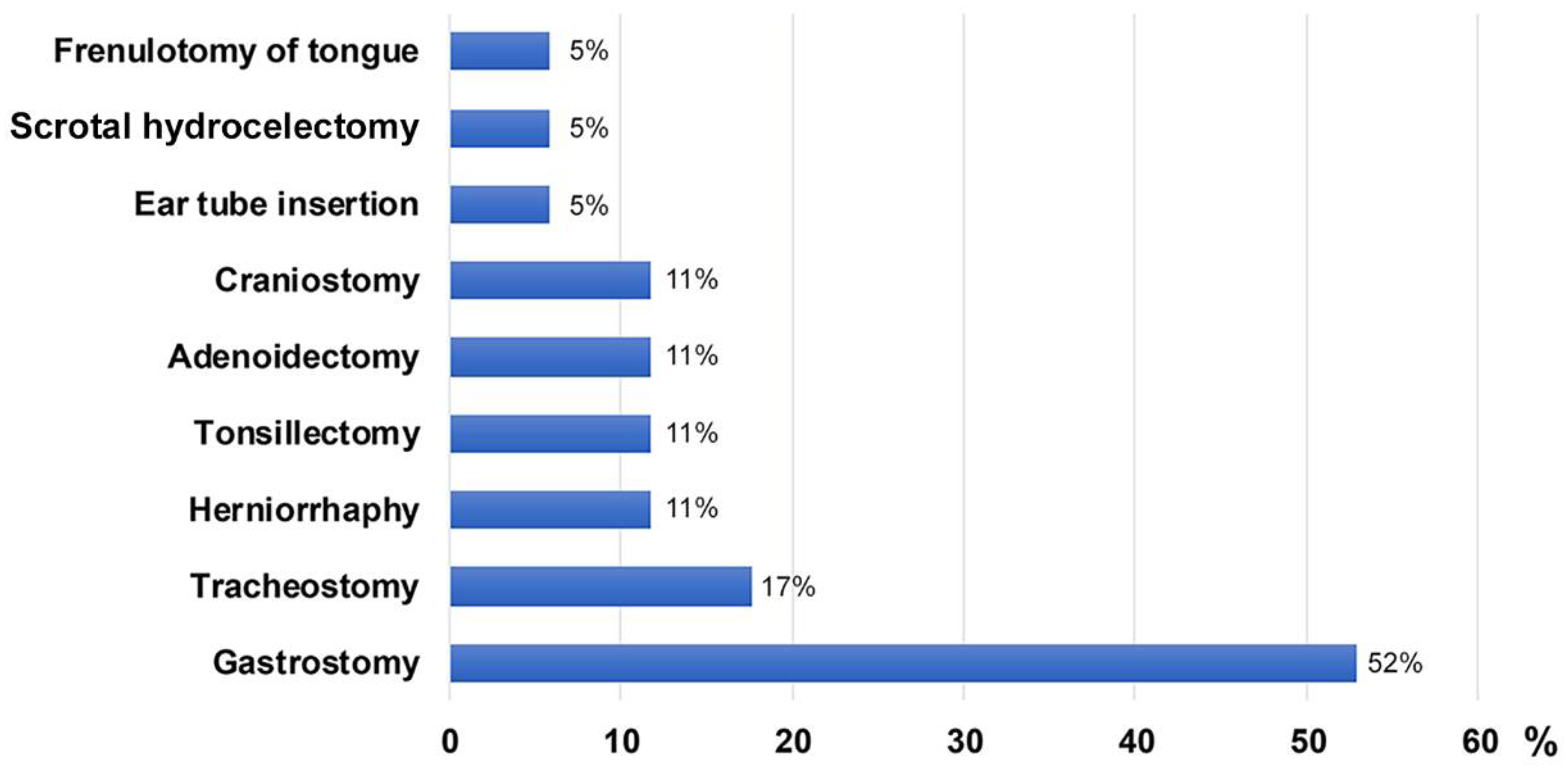
| Gastrostomy | 18 (52.9%) | 9 (50%) | 9 (64.3%) | 0.65 | 0 |
| Age at gastrostomy (mean years ± SD) | 17.2 ± 1.9 | 16.8 ± 2.2 | 17.6 ± 1.8 | 0.421 | NA |
| Tracheostomy | 6 (17.6%) | 4 (22.2%) | 2 (14.3%) | 0.67 | 0 |
| Age at tracheostomy (mean years ± SD) | 17.8 ± 3.7 | 16.3 ± 1.3 | 21 ± 5.7 | 0.24 | NA |
| Deafness | 17 (50%) | 11 (64.7%) | 4 (30.8%) | 0.14 | 2 (100%) |
| Bed-ridden status | 21 (61.8%) | 12 (57.1%) | 9 (42.9%) | >0.99 | NA |
| Age at onset of bed-ridden state (mean years ± SD) | 15.1 ± 1.9 | 15.3 ± 1.6 | 14.9 ± 2.4 | 0.667 | NA |
| First Symptom a | Total, N (%) | IIIA, N (%) | IIIB, N (%) | p-Value | IIIC, N (%) | |
|---|---|---|---|---|---|---|
| Language retardation | 30 (88.2) | 18 (100) | 11 (78.6) | 0.073 | 1 (50) | |
| Motor retardation | 26 (76.5) | 11 (61.1) | 14 (100) | 0.010 * | 1 (50) | |
| Language and motor retardation | 22 (64.7) | 11 (61.1) | 11 (78.6) | 0.721 | 0 (0) | |
| Behavioural abnormalities | 14 (41.2) | 8 (44.4) | 6 (42.9) | >0.99 | 0 (0) | |
| Coarse facial features | 6 (17.6) | 3 (16.7) | 2 (14.3) | >0.99 | 1 (50) | |
| Skeletal abnormalities | 4 (11.7) | 2 (16.7) | 1 (7.1) | >0.99 | 1 (50) | |
| Hearing loss | 4 (11.7) | 1 (5.6) | 2 (14.3) | 0.568 | 1 (50) | |
| Associated Symptom a after Diagnosis | N (%) | Median Age, Year (Range) | ||||
| Impairment of speech | 33 (97.1) | 3.9 (1.5–5.0) | ||||
| Behavioural abnormalities | 32 (94.2) | 4.0 (0.5–7.0) | ||||
| Clumsy walking | 31 (91.2) | 10.0 (4.6–15.0) | ||||
| Macrocephaly and coarse face | 30 (88.2) | 3.0 (1.0–4.0) | ||||
| Sleep disorder | 30 (88.2) | 3.7 (0.5–6.6) | ||||
| Hirsutism | 29 (85.3) | |||||
| Mongolian spots | 22 (64.7) | |||||
| Hearing loss | 17 (50.0) | 5.0 (0–8.5) | ||||
| Recurrent otitis | 13 (38.2) | 2.5 (1.0–4.0) | ||||
| Recurrent diarrhoea | 8 (23.5) | 3.5 (0.5–5.5) | ||||
| Hernia | 8 (23.5) | 0.6 (0–1.2) | ||||
| Characteristic | Survivor (N = 21) | Non-Survivor (N = 13) | p-Value |
|---|---|---|---|
| Sex | >0.99 | ||
| Male | 12(57.2%) | 8 (61.5%) | |
| Female | 9 (42.8%) | 5 (38.5%) | |
| Type | 0.68 | ||
| IIIA | 12 (57.2%) | 6 (46.2%) | |
| IIIB | 7 (33.3%) | 7 (53.8%) | |
| IIIC | 2 (9.5%) | 0 (0%) | |
| Age at death (mean years ± SD) | 14.4 ± 4.1 | ||
| Age at diagnosis (mean years ± SD) | 5.6 ± 2.3 | 7.3 ± 1.8 | 0.029 * |
| Age at symptom onset (mean years ± SD) | 2.8 ± 0.8 | 2.9 ± 0.9 | 0.69 |
| Lag time to diagnosis (mean years ± SD) | 2.8 ± 1.8 | 4.8 ± 3.0 | 0.045 * |
| Cardiac evaluation by TTE | 11 (100%) | 8 (100%) | |
| MR | 8 (72.7%) | 7 (87.5%) | 0.60 |
| TR | 9 (81.8%) | 7 (87.5%) | >0.99 |
| MV thickening | 6 (54.6%) | 7 (87.5%) | 0.18 |
| Abdominal US | 14 (100%) | 7 (100%) | |
| Hepatosplenomegaly | 11 (78.6%) | 6 (85.7%) | >0.99 |
| SMA syndrome | 2 (14.3%) | 4 (57.1%) | 0.52 |
| Clinical findings | |||
| History of AEDs medication | 13 (61.9%) | 13 (100%) | 0.027 * |
| Gastrostomy | 9 (42.9%) | 9 (69.2%) | 0.31 |
| Tracheostomy | 4 (19%) | 2 (15.4%) | >0.99 |
| Deafness | 12 (57.1%) | 5 (45.5%) | >0.99 |
| Bed-ridden status | 10 (47.6%) | 11 (78.6%) | 0.26 |
| Age of Death (Years) | Reference | Study Disease | Number of Patients | Follow-Up Period |
|---|---|---|---|---|
| 18.5 | Lin et al. [27] | MPS IIIA, IIIB, IIIC | 15 | 35 years |
| 16.2 | Malm et al. [26] | MPS IIIA | 22 | 30 years |
| 15.2 | Meyer et al. [18] | MPS IIIA, IIIB | 10 | 3 years |
| 17 (60% probability) | Buhrman et al. [12] | MPS IIIA | 46 | 13 years |
| 15 | Delgadillo et al. [17] | MPS IIIA | 10 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-S.; Yang, A.; Noh, E.-s.; Kim, C.; Bae, G.Y.; Lim, H.H.; Park, H.-D.; Cho, S.Y.; Jin, D.-K. Natural History and Molecular Characteristics of Korean Patients with Mucopolysaccharidosis Type III. J. Pers. Med. 2022, 12, 665. https://doi.org/10.3390/jpm12050665
Kim M-S, Yang A, Noh E-s, Kim C, Bae GY, Lim HH, Park H-D, Cho SY, Jin D-K. Natural History and Molecular Characteristics of Korean Patients with Mucopolysaccharidosis Type III. Journal of Personalized Medicine. 2022; 12(5):665. https://doi.org/10.3390/jpm12050665
Chicago/Turabian StyleKim, Min-Sun, Aram Yang, Eu-seon Noh, Chiwoo Kim, Ga Young Bae, Han Hyuk Lim, Hyung-Doo Park, Sung Yoon Cho, and Dong-Kyu Jin. 2022. "Natural History and Molecular Characteristics of Korean Patients with Mucopolysaccharidosis Type III" Journal of Personalized Medicine 12, no. 5: 665. https://doi.org/10.3390/jpm12050665
APA StyleKim, M.-S., Yang, A., Noh, E.-s., Kim, C., Bae, G. Y., Lim, H. H., Park, H.-D., Cho, S. Y., & Jin, D.-K. (2022). Natural History and Molecular Characteristics of Korean Patients with Mucopolysaccharidosis Type III. Journal of Personalized Medicine, 12(5), 665. https://doi.org/10.3390/jpm12050665