
WGCNA-Based DNA Methylation Profiling Analysis on Allopurinol-Induced Severe Cutaneous Adverse Reactions: A DNA Methylation Signature for Predisposing Drug Hypersensitivity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Blood Sample Collection, Genomic DNA Extraction, and Methylation Beadchip Assay
2.3. Analysis Software
2.4. Screening Differentially Methylated Positions and Regions
2.5. Algorithm of Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. The Definition of Dissimilarity Measurement in WGCNA
3. Results
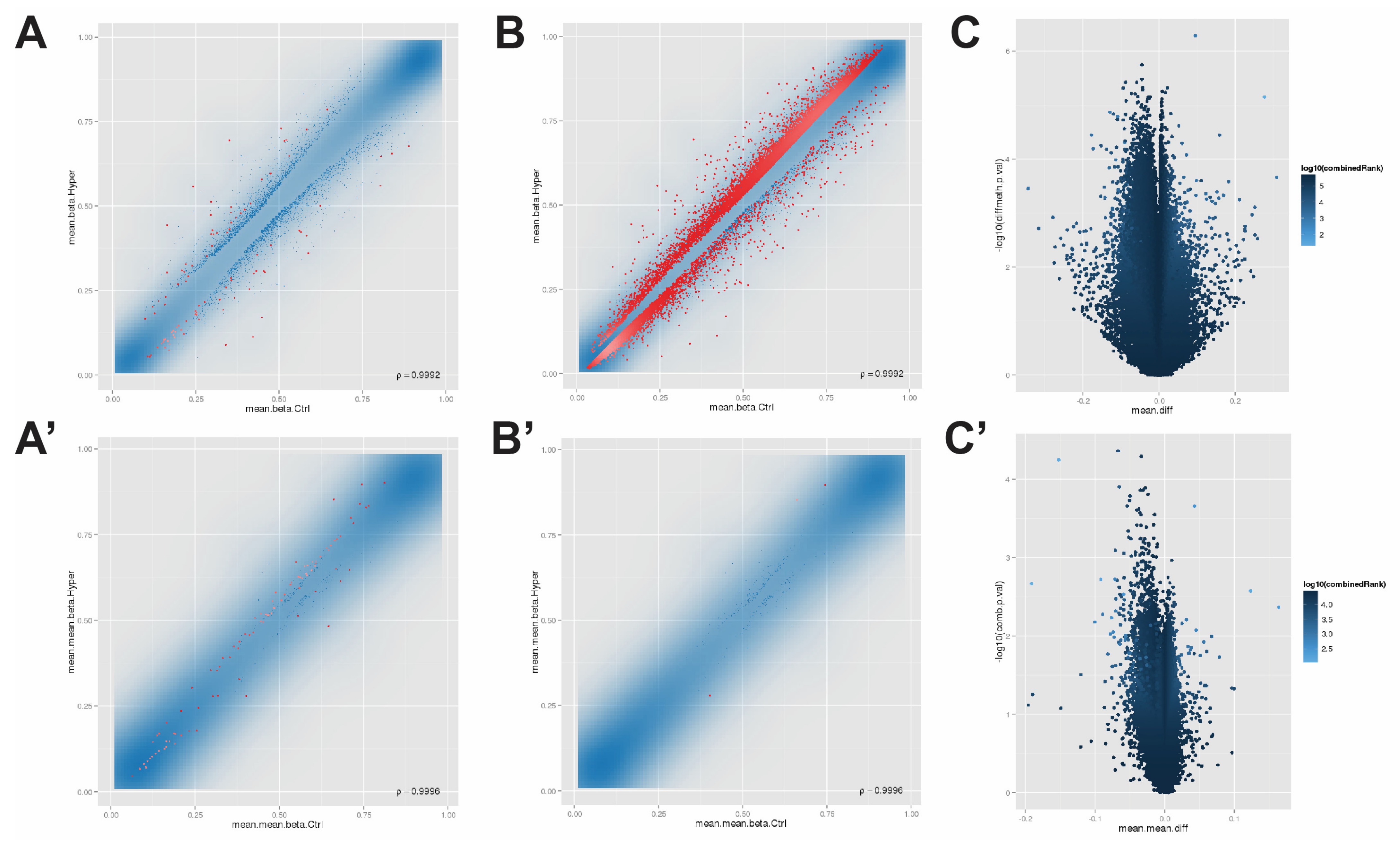
3.1. The Methylation Array Data Were Quality Controlled and Normalized
3.2. Screening Differentially Methylated Positions and Regions
3.3. Selection of Parameters for WGCNA
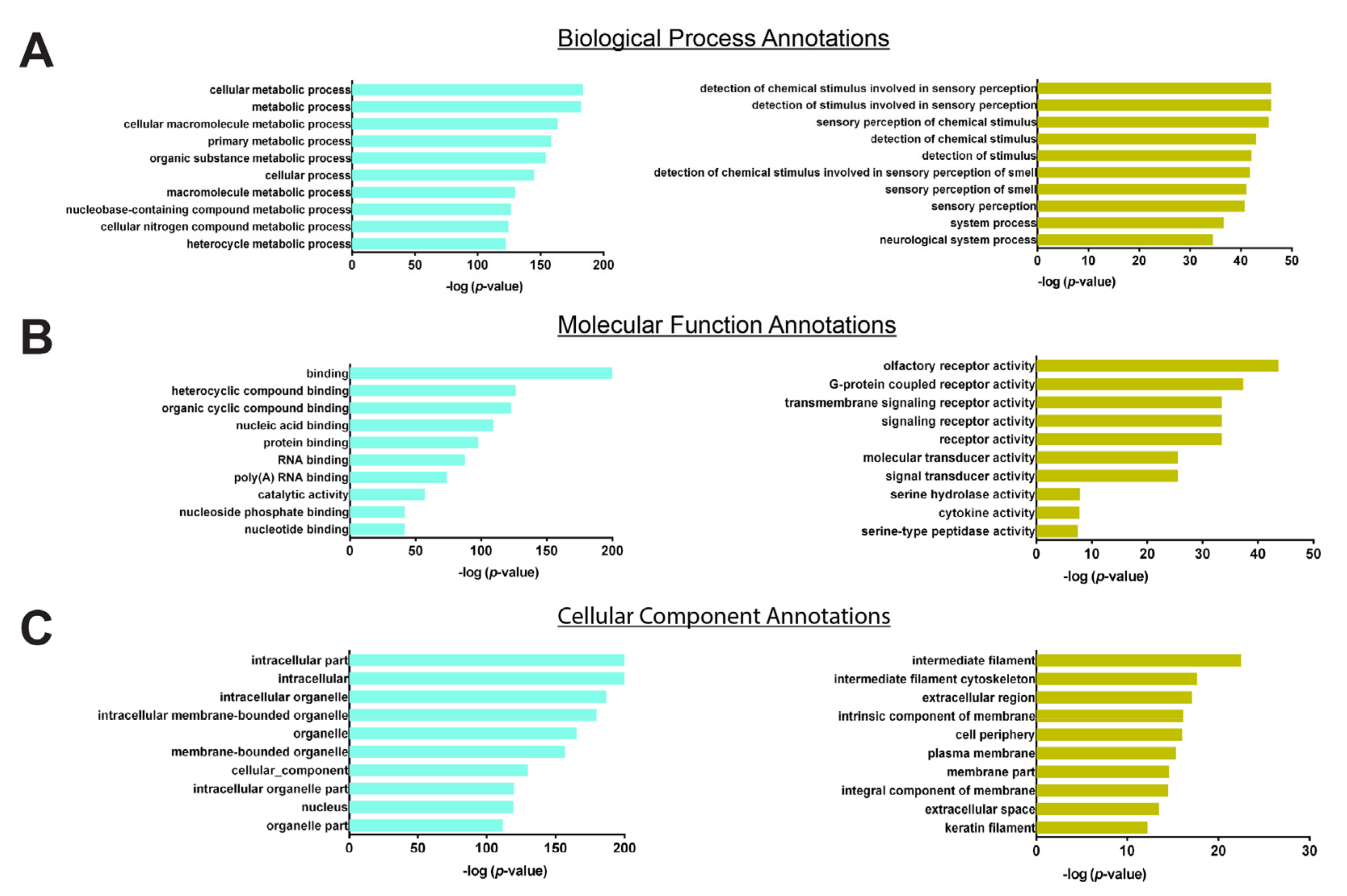
3.4. Hub Genes Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, L.; Xiong, Y.; Qin, C.; Zhang, W.; Chen, X.; Li, J.; Zhou, H. HLA-B*5801 is strongly associated with allopurinol-induced severe cutaneous adverse reactions in Han Chinese: A multicenter retrospective case-control clinical study. Br. J. Dermatol. 2015, 173, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.H.; Hung, S.I. Genetic markers and danger signals in Stevens-Johnson syndrome and toxic epidermal necrolysis. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2010, 59, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.H.; Hung, S.I.; Yang, J.Y.; Su, S.C.; Huang, S.P.; Wei, C.Y.; Chin, S.W.; Chiou, C.C.; Chu, S.C.; Ho, H.C.; et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat. Med. 2008, 14, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L. Current pharmacogenetic perspective on Stevens-Johnson syndrome and toxic epidermal necrolysis. Front. Pharmacol. 2021, 12, 588063. [Google Scholar] [CrossRef] [PubMed]
- Renaudineau, Y.; Youinou, P. Epigenetics and autoimmunity, with special emphasis on methylation. Keio J. Med. 2011, 60, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Meda, F.; Folci, M.; Baccarelli, A.; Selmi, C. The epigenetics of autoimmunity. Cell. Mol. Immunol. 2011, 8, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M.; Mabert, K.; Rauen, T.; Tsokos, G.C. DNA methylation in systemic lupus erythematosus. Epigenomics 2017, 9, 505–525. [Google Scholar] [CrossRef] [PubMed]
- Nemtsova, M.V.; Zaletaev, D.V.; Bure, I.V.; Mikhaylenko, D.S.; Kuznetsova, E.B.; Alekseeva, E.A.; Beloukhova, M.I.; Deviatkin, A.A.; Lukashev, A.N.; Zamyatnin, A.A., Jr. Epigenetic changes in the pathogenesis of rheumatoid arthritis. Front. Genet. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.S. Epigenetics in multiple sclerosis. Adv. Exp. Med. Biol. 2020, 1253, 309–374. [Google Scholar] [PubMed]
- Monroy-Arreola, A.; Duran-Figueroa, N.V.; Mendez-Flores, S.; Dominguez-Cherit, J.; Watkinson, J.; Badillo-Corona, J.A.; Whitaker, P.; Naisbitt, D.J.; Castrejon-Flores, J.L. Up-regulation of T-cell activation micrornas in drug-specific CD4(+) T-cells from hypersensitive patients. Chem. Res. Toxicol. 2018, 31, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Elbakkoush, A.A.; Khaleel, A.; Liu, C.T. MicroRNA and gene signature of severe cutaneous drug hypersensitivity reactions reveal the role of miR-483-5p/miR-28-5p in inflammation by targeting granulysin gene. Trop. J. Pharm. Res. 2017, 16, 771–779. [Google Scholar] [CrossRef][Green Version]
- Ichihara, A.; Wang, Z.; Jinnin, M.; Izuno, Y.; Shimozono, N.; Yamane, K.; Fujisawa, A.; Moriya, C.; Fukushima, S.; Inoue, Y.; et al. Upregulation of miR-18a-5p contributes to epidermal necrolysis in severe drug eruptions. J. Allergy Clin. Immunol. 2014, 133, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Webb, S.; Kerr, A.R.; Illingworth, R.S.; Guy, J.; Andrews, R.; Bird, A. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011, 21, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Van Emburgh, B.O.; Robertson, K.D. DNA methylation in development and human disease. Mutat. Res. 2008, 647, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Roujeau, J.C. Clinical heterogeneity of drug hypersensitivity. Toxicology 2005, 209, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Cheng, L.; Xiong, Y.; Hu, L.; Luo, Z.; Zhou, M.; Li, J.; Xie, H.; He, F.; Yuan, X.; et al. PSORS1C1 hypomethylation is associated with allopurinol-induced severe cutaneous adverse reactions during disease onset period: A multicenter retrospective case-control clinical study in Han Chinese. Front. Pharmacol. 2017, 8, 923. [Google Scholar] [CrossRef] [PubMed]
- Illumina. Infinium Humanmethylation450 Beadchip Kit Introduction. Available online: http://www.illumina.com/products/methylation_450_beadchip_kits.html (accessed on 20 October 2021).
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yan, L.; Hu, Q.; Sucheston, L.E.; Higgins, M.J.; Ambrosone, C.B.; Johnson, C.S.; Smiraglia, D.J.; Liu, S. IMA: An R package for high-throughput analysis of Illumina’s 450k infinium methylation data. Bioinformatics 2012, 28, 729–730. [Google Scholar] [CrossRef] [PubMed]
- Warden, C.D.; Lee, H.; Tompkins, J.D.; Li, X.; Wang, C.; Riggs, A.D.; Yu, H.; Jove, R.; Yuan, Y.C. COHCAP: An integrative genomic pipeline for single-nucleotide resolution DNA methylation analysis. Nucleic Acids Res. 2013, 41, e117. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Muller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of beta-value and m-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Horvath, S. Understanding network concepts in modules. BMC Syst Biol. 2007, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Yip, A.M.; Horvath, S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinform. 2007, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed]
- Weisstein, E.W. “Logit Transformation.” From MathWorld—A Wolfram Web Resource. Available online: https://mathworld.wolfram.com/LogitTransformation.html (accessed on 20 October 2021).
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [PubMed]
- Fuller, T.F.; Ghazalpour, A.; Aten, J.E.; Drake, T.A.; Lusis, A.J.; Horvath, S. Weighted gene co-expression network analysis strategies applied to mouse weight. Mamm. Genome 2007, 18, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Hieke, N.; Loffler, A.S.; Kaizuka, T.; Berleth, N.; Bohler, P.; Driessen, S.; Stuhldreier, F.; Friesen, O.; Assani, K.; Schmitz, K.; et al. Expression of a ULK1/2 binding-deficient ATG13 variant can partially restore autophagic activity in ATG13-deficient cells. Autophagy 2015, 11, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Mizushima, N. Atg13 is essential for autophagy and cardiac development in mice. Mol. Cell. Biol. 2016, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Vernia, S.; Heredia, M.; Criado, O.; Rodriguez de Cordoba, S.; Garcia-Roves, P.M.; Cansell, C.; Denis, R.; Luquet, S.; Foufelle, F.; Ferre, P.; et al. Laforin, a dual specificity phosphatase involved in Lafora disease, regulates insulin response and whole-body energy balance in mice. Hum. Mol. Genet. 2011, 20, 2571–2584. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Tiberia, E.; Pereira, S.; Zhao, X.; Pencea, N.; Wheeler, A.L.; Yu, W.Q.; Ivovic, A.; Naranian, T.; Israelian, N.; et al. Deficiency of a glycogen synthase-associated protein, Epm2aip1, causes decreased glycogen synthesis and hepatic insulin resistance. J. Biol. Chem. 2013, 288, 34627–34637. [Google Scholar] [CrossRef] [PubMed]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013, 73, 2817–2828. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wen, J.; Chang, C.C.; Zhou, X. Discovering transcription and splicing networks in myelodysplastic syndromes. PLoS ONE 2013, 8, e79118. [Google Scholar] [CrossRef] [PubMed]
- Raihan, O.; Brishti, A.; Li, Q.; Zhang, Q.; Li, D.; Li, X.; Zhang, Q.; Xie, Z.; Li, J.; Zhang, J.; et al. SFRS11 loss leads to aging-associated cognitive decline by modulating LRP8 and ApoE. Cell Rep. 2019, 28, 78–90.e76. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.J.; Lombroso, S.I.; Fischer, D.K.; Carpenter, M.D.; Marchione, D.M.; Hamilton, P.J.; Lim, C.J.; Neve, R.L.; Garcia, B.A.; Wimmer, M.E.; et al. Chromatin-mediated alternative splicing regulates cocaine-reward behavior. Neuron 2021, 109, 2943–2966.e2948. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. Roles of the transcription factor p53 in keratinocyte carcinomas. Br. J. Dermatol. 2006, 154 (Suppl. 1), 8–10. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, L.; Sun, B.; Xiong, Y.; Hu, L.; Gao, L.; Li, J.; Xie, H.; Chen, X.; Zhang, W.; Zhou, H.-H. WGCNA-Based DNA Methylation Profiling Analysis on Allopurinol-Induced Severe Cutaneous Adverse Reactions: A DNA Methylation Signature for Predisposing Drug Hypersensitivity. J. Pers. Med. 2022, 12, 525. https://doi.org/10.3390/jpm12040525
Cheng L, Sun B, Xiong Y, Hu L, Gao L, Li J, Xie H, Chen X, Zhang W, Zhou H-H. WGCNA-Based DNA Methylation Profiling Analysis on Allopurinol-Induced Severe Cutaneous Adverse Reactions: A DNA Methylation Signature for Predisposing Drug Hypersensitivity. Journal of Personalized Medicine. 2022; 12(4):525. https://doi.org/10.3390/jpm12040525
Chicago/Turabian StyleCheng, Lin, Bao Sun, Yan Xiong, Lei Hu, Lichen Gao, Ji Li, Hongfu Xie, Xiaoping Chen, Wei Zhang, and Hong-Hao Zhou. 2022. "WGCNA-Based DNA Methylation Profiling Analysis on Allopurinol-Induced Severe Cutaneous Adverse Reactions: A DNA Methylation Signature for Predisposing Drug Hypersensitivity" Journal of Personalized Medicine 12, no. 4: 525. https://doi.org/10.3390/jpm12040525
APA StyleCheng, L., Sun, B., Xiong, Y., Hu, L., Gao, L., Li, J., Xie, H., Chen, X., Zhang, W., & Zhou, H.-H. (2022). WGCNA-Based DNA Methylation Profiling Analysis on Allopurinol-Induced Severe Cutaneous Adverse Reactions: A DNA Methylation Signature for Predisposing Drug Hypersensitivity. Journal of Personalized Medicine, 12(4), 525. https://doi.org/10.3390/jpm12040525