
From Negative to Positive Diagnosis: Structural Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene

 , ,
, ,  ,
,
Abstract
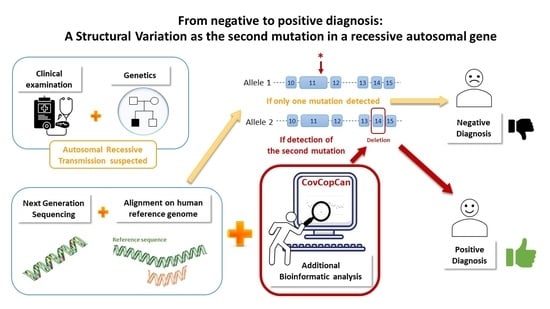
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Next-Generation Sequencing (NGS) and Bioinformatics Analysis
2.3. Verification of Mutations
3. Results
3.1. Patient’s Clinical Description
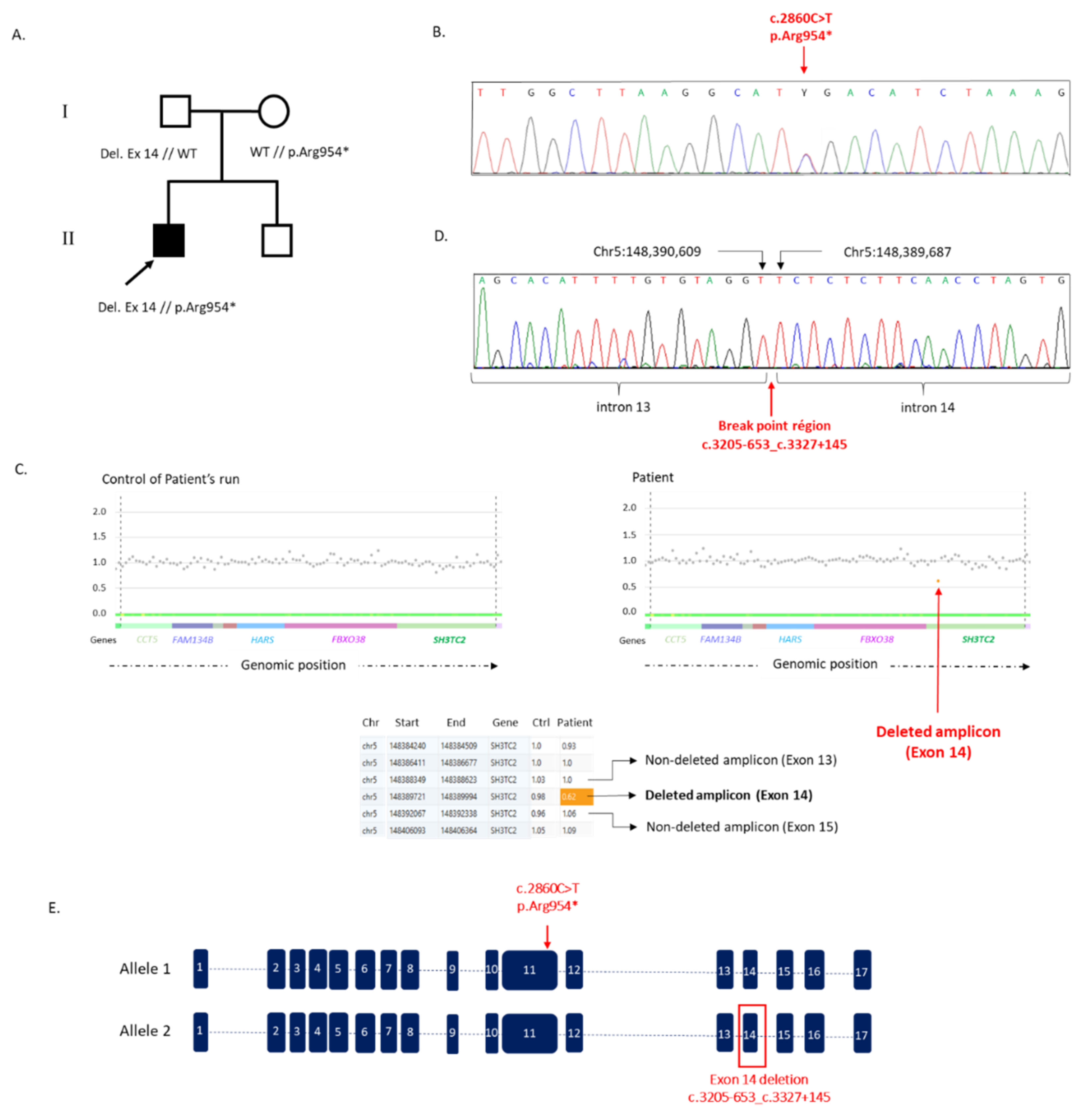
3.2. Detection of SNPs and Structural Variants
3.3. Confirmation of the Structural Variant and of the Nonsense Mutation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Pfarr, N.; Stenzinger, A.; Treue, D.; Endris, V.; Ismaeel, F.; Bangemann, N.; Blohmer, J.U.; Dietel, M.; Loibl, S.; et al. Ioncopy: A novel method for calling copy number alterations in amplicon sequencing data including significance assessment. Oncotarget 2016, 7, 13236–13247. [Google Scholar] [CrossRef] [PubMed]
- Derouault, P.; Parfait, B.; Moulinas, R.; Barrot, C.C.; Sturtz, F.; Merillou, S.; Lia, A.S. ‘COV’COP’ allows to detect CNVs responsible for inherited diseases among amplicons sequencing data. Bioinformatics 2017, 33, 1586–1588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Derouault, P.; Chauzeix, J.; Rizzo, D.; Miressi, F.; Magdelaine, C.; Bourthoumieu, S.; Durand, K.; Dzugan, H.; Feuillard, J.; Sturtz, F.; et al. CovCopCan: An Efficient Tool to Detect Copy Number Variation from Amplicon Sequencing Data in Inherited Diseases and Cancer. PLoS Comput. Biol. 2020, 16, e1007503. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Nam, S.H.; Park, K.S.; Kim, Y.; Kim, J.W.; Lee, E.; Ko, J.M.; Lee, K.A.; Park, I. DeviCNV: Detection and visualization of exon-level copy number variants in targeted next-generation sequencing data. BMC Bioinform. 2018, 19, 381. [Google Scholar] [CrossRef]
- Bird, T.D. Charcot-Marie-Tooth (CMT) Hereditary Neuropathy Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA Duplication Associated with Charcot-Marie-Tooth Disease Type 1. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Matsunami, N.; Smith, B.; Ballard, L.; Lensch, M.W.; Robertson, M.; Albertsen, H.; Hanemann, C.O.; Müller, H.W.; Bird, T.D.; White, R.; et al. Peripheral Myelin Protein-22 Gene Maps in the Duplication in Chromosome 17p11.2 Associated with Charcot-Marie-Tooth 1A. Nat. Genet. 1992, 1, 176–179. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Bolhuis, P.A.; Zorn, I.; Hoogendijk, J.E.; van den Bosch, N.; Hensels, G.W.; Stanton, V.P., Jr.; Housman, D.E.; Fischbeck, K.H.; Ross, D.A.; et al. The Peripheral Myelin Gene PMP-22/GAS-3 is Duplicated in Charcot-Marie-Tooth Disease Type 1A. Nat. Genet. 1992, 1, 166–170. [Google Scholar] [CrossRef]
- Timmerman, V.; Nelis, E.; Van Hul, W.; Nieuwenhuijen, B.W.; Chen, K.L.; Wang, S.; Ben Othman, K.; Cullen, B.; Leach, R.J.; Hanemann, C.O.; et al. The Peripheral Myelin Protein Gene PMP–22 Is Contained within the Charcot–Marie–Tooth Disease Type 1A Duplication. Nat. Genet. 1992, 1, 171–175. [Google Scholar] [CrossRef]
- Murphy, S.M.; Laura, M.; Fawcett, K.; Pandraud, A.; Liu, Y.T.; Davidson, G.L.; Rossor, A.M.; Polke, J.M.; Castleman, V.; Manji, H.; et al. Charcot-Marie-Tooth Disease: Frequency of Genetic Subtypes and Guidelines for Genetic Testing. J. Neurol. Neurosurg. Psychiatry 2012, 83, 706–710. [Google Scholar] [CrossRef]
- Mortreux, J.; Bacquet, J.; Boyer, A.; Alazard, E.; Bellance, R.; Giguet-Valard, A.G.; Cerino, M.; Krahn, M.; Audic, F.; Chabrol, B.; et al. Identification of novel pathogenic copy number variations in Charcot-Marie-Tooth disease. J. Hum. Genet. 2020, 65, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Pyromali, I.; Perani, A.; Nizou, A.; Benslimane, N.; Derouault, P.; Bourthoumieu, S.; Fradin, M.; Sole, G.; Duval, F.; Gomes, C.; et al. New structural variations responsible for Charcot-Marie-Tooth disease: The first two large KIF5A deletions detected by CovCopCan software. Comput. Struct. Biotechnol. J. 2021, 19, 4265–4272. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Bergmann, C.; Stendel, C.; Kirfel, J.; Verpoorten, N.; De Jonghe, P.; Timmerman, V.; Chrast, R.; Verheijen, M.H.G.; Lemke, G.; et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am. J. Hum. Genet. 2003, 73, 1106–1119. [Google Scholar] [CrossRef]
- Arnaud, E.; Zenker, J.; de Preux Charles, A.S.; Stendel, C.; Roos, A.; Médard, J.J.; Tricaud, N.; Kleine, H.; Luscher, B.; Weis, J.; et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 17528–17533. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C.; Peden, A.A.; Buss, F.; Bright, N.A.; Latouche, M.; Reilly, M.M.; Kendrick-Jones, J.; Luzio, J.P. Mistargeting of SH3TC2 away from the recycling endosome causes Charcot-Marie-Tooth disease type 4C. Hum. Mol. Genet. 2010, 19, 1009–1018. [Google Scholar] [CrossRef]
- Azzedine, H.; Salih, M.A. SH3TC2-Related Hereditary Motor and Sensory Neuropathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; Internet; Updated 11 March 2021; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lerat, J.; Magdelaine, C.; Lunati, A.; Dzugan, H.; Dejoie, C.; Rego, M.; Beze Beyrie, P.; Bieth, E.; Calvas, P.; Cintas, P.; et al. Implication of the SH3TC2 gene in Charcot-Marie-Tooth disease associated with deafness and/or scoliosis: Illustration with four new pathogenic variants. J. Neurol. Sci. 2019, 406, 116376. [Google Scholar] [CrossRef]
- Fridman, V.; Bundy, B.; Reilly, M.M.; Pareyson, D.; Bacon, C.; Burns, J.; Day, J.; Feely, S.; Finkel, R.S.; Grider, T.; et al. Inherited Neuropathies Consortium. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: A cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 873–878. [Google Scholar] [CrossRef]
- Yger, M.; Stojkovic, T.; Tardieu, S.; Maisonobe, T.; Brice, A.; Echaniz-Laguna, A.; Alembik, Y.; Girard, S.; Cazeneuve, C.; Leguern, E.; et al. Characteristics of clinical and electrophysiological pattern of Charcot-Marie-Tooth 4C. J. Peripher. Nerv. Syst. 2012, 17, 112–122. [Google Scholar] [CrossRef]
- Laššuthová, P.; Mazanec, R.; Vondráček, P.; Sišková, D.; Haberlová, J.; Sabová, J.; Seeman, P. High frequency of SH3TC2 mutations in Czech HMSN I patients. Clin. Genet. 2011, 80, 334–345. [Google Scholar] [CrossRef]
- Piscosquito, G.; Saveri, P.; Magri, S.; Ciano, C.; Gandioli, C.; Morbin, M.; Bella, D.D.; Moroni, I.; Taroni, F.; Pareyson, D. Screening for SH3TC2 gene mutations in a series of demyelinating recessive Charcot-Marie-Tooth disease (CMT4). J. Peripher. Nerv. Syst. 2016, 21, 142–149. [Google Scholar] [CrossRef]
- Compston, A. Aids to the investigation of peripheral nerve injuries. Medical Research Council: Nerve Injuries Research Committee. His Majesty’s Stationery Office: 1942; pp. 48 (iii) and 74 figures and 7 diagrams; with aids to the examination of the peripheral nervous system. By Michael O’Brien for the Guarantors of Brain. Saunders Elsevier: 2010; pp. [8] 64 and 94 Figures. Brain 2010, 133, 2838–2844. [Google Scholar] [CrossRef] [PubMed]
- Miressi, F.; Faye, P.A.; Pyromali, I.; Bourthoumieu, S.; Derouault, P.; Husson, M.; Favreau, F.; Sturtz, F.; Magdelaine, C.; Lia, A.S. A mutation can hide another one: Think Structural Variants! Comput. Struct. Biotechnol. J. 2020, 18, 2095–2099. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Andary, M.; Buschbacher, R.; Del Toro, D.; Smith, B.; So, Y.; Zimmermann, K.; Dillingham, T.R. Electrodiagnostic reference values for upper and lower limb nerve conduction studies in adult populations. Muscle Nerve 2016, 54, 371–377. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Motor Nerve Conduction Values (MNCV) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ulnar | Median | Tibial | Fibular Right | Fibular Left | |||||
| Vel (m/s) | Amp (mV) | Vel (m/s) | Amp (mV) | Vel (m/s) | Amp (mV) | Vel (m/s) | Amp (mV) | Vel (m/s) | Amp (mV) |
| 20.7 | 2.9 | 26.2 | 0.97 | NR | NR | 22.1 | 0.23 | 21.0 | 0.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyromali, I.; Benslimane, N.; Favreau, F.; Goizet, C.; Lazaro, L.; Vitry, M.; Derouault, P.; Sturtz, F.; Magdelaine, C.; Lia, A.-S. From Negative to Positive Diagnosis: Structural Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene. J. Pers. Med. 2022, 12, 212. https://doi.org/10.3390/jpm12020212
Pyromali I, Benslimane N, Favreau F, Goizet C, Lazaro L, Vitry M, Derouault P, Sturtz F, Magdelaine C, Lia A-S. From Negative to Positive Diagnosis: Structural Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene. Journal of Personalized Medicine. 2022; 12(2):212. https://doi.org/10.3390/jpm12020212
Chicago/Turabian StylePyromali, Ioanna, Nesrine Benslimane, Frédéric Favreau, Cyril Goizet, Leila Lazaro, Martine Vitry, Paco Derouault, Franck Sturtz, Corinne Magdelaine, and Anne-Sophie Lia. 2022. "From Negative to Positive Diagnosis: Structural Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene" Journal of Personalized Medicine 12, no. 2: 212. https://doi.org/10.3390/jpm12020212
APA StylePyromali, I., Benslimane, N., Favreau, F., Goizet, C., Lazaro, L., Vitry, M., Derouault, P., Sturtz, F., Magdelaine, C., & Lia, A.-S. (2022). From Negative to Positive Diagnosis: Structural Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene. Journal of Personalized Medicine, 12(2), 212. https://doi.org/10.3390/jpm12020212