
Deep Learning-Based Prediction of Molecular Tumor Biomarkers from H&E: A Practical Review


Abstract
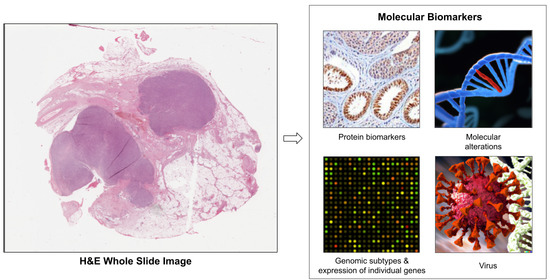
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Deep Learning Methods for Whole Slide Images
2.1. Bottom-Up Approaches to Learning Features
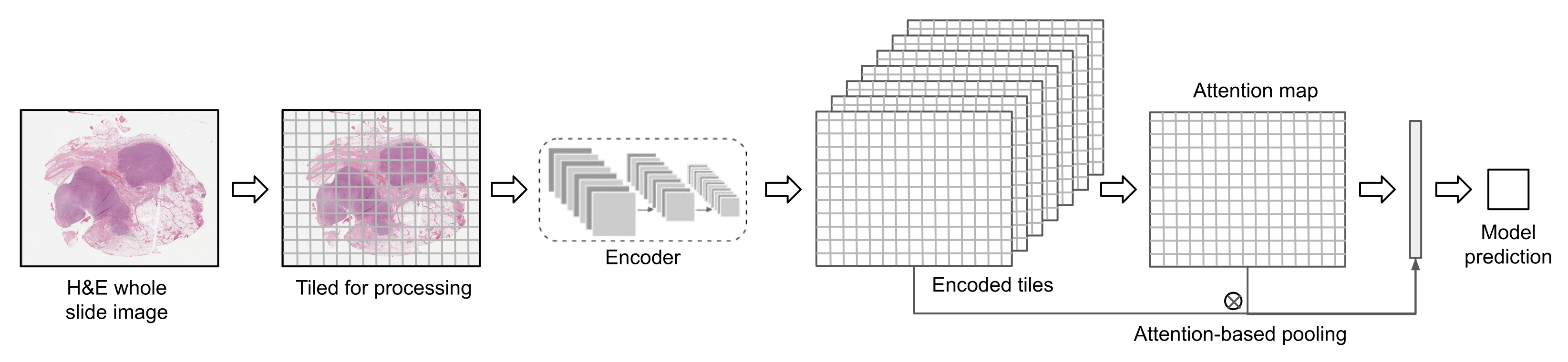
2.1.1. Learning Tile Features
2.1.2. Tile Aggregation
2.1.3. Weakly Supervised Learning with Self-Supervised Features
2.1.4. Histopathology-Based Transfer Learning
2.2. Learning with Pathologist-Driven Features
2.2.1. Hand-Crafted Tissue Features
2.2.2. Hybrid Models
2.3. Strongly Supervised Biomarkers
3. Challenges
3.1. Tile Selection
3.2. Magnification for Bottom-Up Approaches
3.3. Quality Control
3.4. Explainability
3.5. Validation
3.6. Domain Generalizability
3.7. Batch Effects
3.8. Dataset Diversity and Spurious Correlations
3.9. Small Datasets
4. Opportunities
4.1. Biomarker Heterogeneity and Outcomes
4.2. Pan-Cancer Modeling
4.3. Multimodal Models
4.4. New Model Types for WSIs
4.5. Datasets and Challenges
5. Discussion
- Which regions of tissue should be included when training a model?
- What magnification is best?
- What effect do artifacts have? Can they be detected and discarded systematically?
- Can we create more explainable models?
- How thorough do we need to be in validating with external cohorts?
- What batch effects do we need to watch out for?
- How can we be sure that we’ve detected all spurious correlations?
- Can we create models that generalize to different scanners, medical centers, or patient populations?
- How do we mitigate bias?
- Can we train models with a small number of patient samples?
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CNN | convolutional neural network |
| FFPE | formalin-fixed paraffin-embedded |
| GPU | graphics processing unit |
| H&E | hematoxylin and eosin |
| HRD | homologous recombination deficiency |
| IHC | immunohistochemistry |
| MIL | multiple instance learning |
| MSI | microsatellite instability |
| TCGA | The Cancer Genome Atlas |
| WSI | whole slide image |
References
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Couture, H.D. Deep learning-based histology biomarkers: Recent advances and challenges for clinical use. Digit. Pathol. Assoc. 2020. Available online: https://digitalpathologyassociation.org/blog/deep-learning-based-histology-biomarkers-recent-advances-and-challenges-for-clinical-use (accessed on 1 September 2022).
- Jenkins, M.A.; Hayashi, S.; O’shea, A.M.; Burgart, L.J.; Smyrk, T.C.; Shimizu, D.; Waring, P.M.; Ruszkiewicz, A.R.; Pollett, A.F.; Redston, M.; et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: A population-based study. Gastroenterology 2007, 133, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Greenson, J.K.; Huang, S.C.; Herron, C.; Moreno, V.; Bonner, J.D.; Tomsho, L.P.; Ben-Izhak, O.; Cohen, H.I.; Trougouboff, P.; Bejhar, J.; et al. Pathologic predictors of microsatellite instability in colorectal cancer. Am. J. Surg. Pathol. 2009, 33, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.; Kumar, N.; Sethi, A.; Gann, P.H. Detecting multiple sub-types of breast cancer in a single patient. In Proceedings of the 2016 IEEE International Conference on Image Processing (ICIP), Phoenix, AZ, USA, 25–28 September 2016; pp. 2648–2652. [Google Scholar]
- Couture, H.D.; Marron, J.; Thomas, N.E.; Perou, C.M.; Niethammer, M. Hierarchical task-driven feature learning for tumor histology. In Proceedings of the 2015 IEEE 12th International Symposium on Biomedical Imaging (ISBI), Brooklyn, NY, USA, 16–19 April 2015; pp. 999–1003. [Google Scholar]
- Bera, K.; Schalper, K.A.; Rimm, D.L.; Velcheti, V.; Madabhushi, A. Artificial intelligence in digital pathology—new tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 2019, 16, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Shmatko, A.; Ghaffari Laleh, N.; Gerstung, M.; Kather, J.N. Artificial intelligence in histopathology: Enhancing cancer research and clinical oncology. Nat. Cancer 2022, 3, 1026–1038. [Google Scholar] [CrossRef]
- Hildebrand, L.A.; Pierce, C.J.; Dennis, M.; Paracha, M.; Maoz, A. Artificial intelligence for histology-based detection of microsatellite instability and prediction of response to immunotherapy in colorectal cancer. Cancers 2021, 13, 391. [Google Scholar] [CrossRef]
- Echle, A.; Laleh, N.G.; Schrammen, P.L.; West, N.P.; Trautwein, C.; Brinker, T.J.; Gruber, S.B.; Buelow, R.D.; Boor, P.; Grabsch, H.I.; et al. Deep learning for the detection of microsatellite instability from histology images in colorectal cancer: A systematic literature review. ImmunoInformatics 2021, 3, 100008. [Google Scholar] [CrossRef]
- Alam, M.R.; Abdul-Ghafar, J.; Yim, K.; Thakur, N.; Lee, S.H.; Jang, H.J.; Jung, C.K.; Chong, Y. Recent Applications of Artificial Intelligence from Histopathologic Image-Based Prediction of Microsatellite Instability in Solid Cancers: A Systematic Review. Cancers 2022, 14, 2590. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, E.Y.; Luchini, C.; Eccher, A.; Tizaoui, K.; Shin, J.I.; Lim, B.J. Artificial Intelligence for Predicting Microsatellite Instability Based on Tumor Histomorphology: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2462. [Google Scholar] [CrossRef]
- Lee, S.H.; Jang, H.J. Deep learning-based prediction of molecular cancer biomarkers from tissue slides: A new tool for precision oncology. Clin. Mol. Hepatol. 2022, 28, 754–772. [Google Scholar] [CrossRef]
- Cifci, D.; Foersch, S.; Kather, J.N. Artificial intelligence to identify genetic alterations in conventional histopathology. J. Pathol. 2022, 257, 430–444. [Google Scholar] [CrossRef]
- Popovici, V.; Budinská, E.; Dušek, L.; Kozubek, M.; Bosman, F. Image-based surrogate biomarkers for molecular subtypes of colorectal cancer. Bioinformatics 2017, 33, 2002–2009. [Google Scholar] [CrossRef] [Green Version]
- Couture, H.D.; Williams, L.A.; Geradts, J.; Nyante, S.J.; Butler, E.N.; Marron, J.; Perou, C.M.; Troester, M.A.; Niethammer, M. Image analysis with deep learning to predict breast cancer grade, ER status, histologic subtype, and intrinsic subtype. NPJ Breast Cancer 2018, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Park, S.; Lee, S.H.; Hwang, T.H. Using transfer learning on whole slide images to predict tumor mutational burden in bladder cancer patients. bioRxiv 2019, 554527. [Google Scholar]
- Qu, H.; Zhou, M.; Yan, Z.; Wang, H.; Rustgi, V.K.; Zhang, S.; Gevaert, O.; Metaxas, D.N. Genetic mutation and biological pathway prediction based on whole slide images in breast carcinoma using deep learning. NPJ Precis. Oncol. 2021, 5, 1–11. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Yu, K.H.; Wang, F.; Berry, G.J.; Re, C.; Altman, R.B.; Snyder, M.; Kohane, I.S. Classifying non-small cell lung cancer types and transcriptomic subtypes using convolutional neural networks. J. Am. Med. Inform. Assoc. 2020, 27, 757–769. [Google Scholar] [CrossRef]
- Sirinukunwattana, K.; Domingo, E.; Richman, S.; Redmond, K.L.; Blake, A.; Verrill, C.; Leedham, S.J.; Chatzipli, A.; Hardy, C.; Whalley, C.; et al. Image-based consensus molecular subtype classification (imCMS) of colorectal cancer using deep learning. bioRxiv 2019, 645143. [Google Scholar] [CrossRef]
- Kim, R.H.; Nomikou, S.; Coudray, N.; Jour, G.; Dawood, Z.; Hong, R.; Esteva, E.; Sakellaropoulos, T.; Donnelly, D.; Moran, U.; et al. A deep learning approach for rapid mutational screening in melanoma. bioRxiv 2020, 610311. [Google Scholar]
- Kather, J.N.; Pearson, A.T.; Halama, N.; Jäger, D.; Krause, J.; Loosen, S.H.; Marx, A.; Boor, P.; Tacke, F.; Neumann, U.P.; et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 2019, 25, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Schulte, J.; Grabsch, H.I.; Loeffler, C.; Muti, H.; Dolezal, J.; Srisuwananukorn, A.; Agrawal, N.; Kochanny, S.; von Stillfried, S.; et al. Deep learning detects virus presence in cancer histology. bioRxiv 2019, 690206. [Google Scholar]
- Kather, J.N.; Heij, L.R.; Grabsch, H.I.; Loeffler, C.; Echle, A.; Muti, H.S.; Krause, J.; Niehues, J.M.; Sommer, K.A.; Bankhead, P.; et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer 2020, 1, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Koohbanani, N.A.; Unnikrishnan, B.; Khurram, S.A.; Krishnaswamy, P.; Rajpoot, N. Self-path: Self-supervision for classification of pathology images with limited annotations. IEEE Trans. Med. Imaging 2021, 40, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Ciga, O.; Xu, T.; Martel, A.L. Self supervised contrastive learning for digital histopathology. Mach. Learn. Appl. 2022, 7, 100198. [Google Scholar] [CrossRef]
- Fashi, P.A.; Hemati, S.; Babaie, M.; Gonzalez, R.; Tizhoosh, H. A self-supervised contrastive learning approach for whole slide image representation in digital pathology. J. Pathol. Inform. 2022, 13, 100133. [Google Scholar] [CrossRef]
- Rawat, R.R.; Ortega, I.; Roy, P.; Sha, F.; Shibata, D.; Ruderman, D.; Agus, D.B. Deep learned tissue "fingerprints" classify breast cancers by ER/PR/Her2 status from H&E images. Sci. Rep. 2020, 10, 1–13. [Google Scholar]
- Liu, Y.; Wang, W.; Ren, C.X.; Dai, D.Q. MetaCon: Meta Contrastive Learning for Microsatellite Instability Detection. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention; Springer: Berlin/Heidelberg, Germany, 2021; pp. 267–276. [Google Scholar]
- Guo, B.; Jonnagaddala, J.; Zhang, H.; Xu, X.S. Predicting microsatellite instability and key biomarkers in colorectal cancer from H&E-stained images: Achieving SOTA with Less Data using Swin Transformer. arXiv 2022, arXiv:2208.10495. [Google Scholar]
- Fu, Y.; Jung, A.W.; Torne, R.V.; Gonzalez, S.; Vöhringer, H.; Shmatko, A.; Yates, L.R.; Jimenez-Linan, M.; Moore, L.; Gerstung, M. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat. Cancer 2020, 1, 800–810. [Google Scholar] [CrossRef]
- Loeffler, C.M.L.; Bruechle, N.O.; Jung, M.; Seillier, L.; Rose, M.; Laleh, N.G.; Knuechel, R.; Brinker, T.J.; Trautwein, C.; Gaisa, N.T.; et al. Artificial Intelligence–based Detection of FGFR3 Mutational Status Directly from Routine Histology in Bladder Cancer: A Possible Preselection for Molecular Testing? Eur. Urol. Focus 2022, 8, 472–479. [Google Scholar] [CrossRef]
- Xu, Z.; Verma, A.; Naveed, U.; Bakhoum, S.F.; Khosravi, P.; Elemento, O. Deep learning predicts chromosomal instability from histopathology images. IScience 2021, 24, 102394. [Google Scholar] [CrossRef]
- Jang, H.J.; Lee, A.; Kang, J.; Song, I.H.; Lee, S.H. Prediction of genetic alterations from gastric cancer histopathology images using a fully automated deep learning approach. World J. Gastroenterol. 2021, 27, 7687. [Google Scholar] [CrossRef]
- Ho, D.J.; Chui, M.H.; Vanderbilt, C.M.; Jung, J.; Robson, M.E.; Park, C.S.; Roh, J.; Fuchs, T.J. Deep Interactive Learning-based ovarian cancer segmentation of H&E-stained whole slide images to study morphological patterns of BRCA mutation. arXiv 2022, arXiv:2203.15015. [Google Scholar]
- La Barbera, D.; Polónia, A.; Roitero, K.; Conde-Sousa, E.; Della Mea, V. Detection of her2 from haematoxylin-eosin slides through a cascade of deep learning classifiers via multi-instance learning. J. Imaging 2020, 6, 82. [Google Scholar] [CrossRef]
- Couture, H.D.; Marron, J.S.; Perou, C.M.; Troester, M.A.; Niethammer, M. Multiple instance learning for heterogeneous images: Training a cnn for histopathology. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention; Springer: Berlin/Heidelberg, Germany, 2018; pp. 254–262. [Google Scholar]
- Valieris, R.; Amaro, L.; Osório, C.A.B.d.T.; Bueno, A.P.; Rosales Mitrowsky, R.A.; Carraro, D.M.; Nunes, D.N.; Dias-Neto, E.; Silva, I.T.d. Deep learning predicts underlying features on pathology images with therapeutic relevance for breast and gastric cancer. Cancers 2020, 12, 3687. [Google Scholar] [CrossRef]
- Saillard, C.; Dehaene, O.; Marchand, T.; Moindrot, O.; Kamoun, A.; Schmauch, B.; Jegou, S. Self supervised learning improves dMMR/MSI detection from histology slides across multiple cancers. arXiv 2021, arXiv:2109.05819. [Google Scholar]
- Bilal, M.; Raza, S.E.A.; Azam, A.; Graham, S.; Ilyas, M.; Cree, I.A.; Snead, D.; Minhas, F.; Rajpoot, N.M. Novel deep learning algorithm predicts the status of molecular pathways and key mutations in colorectal cancer from routine histology images. medRxiv 2021. [Google Scholar]
- Höhne, J.; de Zoete, J.; Schmitz, A.A.; Bal, T.; di Tomaso, E.; Lenga, M. Detecting genetic alterations in BRAF and NTRK as oncogenic drivers in digital pathology images: Towards model generalization within and across multiple thyroid cohorts. In Proceedings of the MICCAI Workshop on Computational Pathology, PMLR, Strasbourg, France, 27 September 2021; pp. 105–116. [Google Scholar]
- Abbasi-Sureshjani, S.; Yüce, A.; Schönenberger, S.; Skujevskis, M.; Schalles, U.; Gaire, F.; Korski, K. Molecular subtype prediction for breast cancer using H&E specialized backbone. In Proceedings of the MICCAI Workshop on Computational Pathology, PMLR, Strasbourg, France, 27 September 2021; pp. 1–9. [Google Scholar]
- Schirris, Y.; Gavves, E.; Nederlof, I.; Horlings, H.M.; Teuwen, J. DeepSMILE: Self-supervised heterogeneity-aware multiple instance learning for DNA damage response defect classification directly from H&E whole-slide images. arXiv 2021, arXiv:2107.09405. [Google Scholar]
- Anand, D.; Yashashwi, K.; Kumar, N.; Rane, S.; Gann, P.; Sethi, A. Weakly supervised learning on unannotated hematoxylin and eosin stained slides predicts BRAF mutation in thyroid cancer with high accuracy. J. Pathol. 2021, 255, 232–242. [Google Scholar] [CrossRef]
- Tavolara, T.E.; Niazi, M.; Gower, A.C.; Ginese, M.; Beamer, G.; Gurcan, M.N. Deep learning predicts gene expression as an intermediate data modality to identify susceptibility patterns in Mycobacterium tuberculosis infected Diversity Outbred mice. EBioMedicine 2021, 67, 103388. [Google Scholar] [CrossRef]
- Graziani, M.; Marini, N.; Deutschmann, N.; Janakarajan, N.; Müller, H.; Martínez, M.R. Attention-based Interpretable Regression of Gene Expression in Histology. arXiv 2022, arXiv:2208.13776. [Google Scholar]
- Campanella, G.; Ho, D.; Häggström, I.; Becker, A.S.; Chang, J.; Vanderbilt, C.; Fuchs, T.J. H&E-based Computational Biomarker Enables Universal EGFR Screening for Lung Adenocarcinoma. arXiv 2022, arXiv:2206.10573. [Google Scholar]
- Ilse, M.; Tomczak, J.; Welling, M. Attention-based deep multiple instance learning. In Proceedings of the International Conference on Machine Learning, PMLR, Stockholm, Sweden, 10–15 July 2018; pp. 2127–2136. [Google Scholar]
- Zeng, Q.; Klein, C.; Caruso, S.; Maille, P.; Laleh, N.G.; Sommacale, D.; Laurent, A.; Amaddeo, G.; Gentien, D.; Rapinat, A.; et al. Artificial intelligence predicts immune and inflammatory gene signatures directly from hepatocellular carcinoma histology. J. Hepatol. 2022, 77, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Laleh, N.G.; Muti, H.S.; Loeffler, C.M.L.; Echle, A.; Saldanha, O.L.; Mahmood, F.; Lu, M.Y.; Trautwein, C.; Langer, R.; Dislich, B.; et al. Benchmarking weakly-supervised deep learning pipelines for whole slide classification in computational pathology. Med. Image Anal. 2022, 79, 102474. [Google Scholar] [CrossRef] [PubMed]
- Weitz, P.; Wang, Y.; Hartman, J.; Rantalainen, M. An investigation of attention mechanisms in histopathology whole-slide-image analysis for regression objectives. In Proceedings of the IEEE/CVF International Conference on Computer Vision, Montreal, QC, Canada, 10–17 October 2021; pp. 611–619. [Google Scholar]
- Schmauch, B.; Romagnoni, A.; Pronier, E.; Saillard, C.; Maillé, P.; Calderaro, J.; Kamoun, A.; Sefta, M.; Toldo, S.; Zaslavskiy, M.; et al. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Levy-Jurgenson, A.; Tekpli, X.; Kristensen, V.N.; Yakhini, Z. Spatial transcriptomics inferred from pathology whole-slide images links tumor heterogeneity to survival in breast and lung cancer. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Kartasalo, K.; Valkonen, M.; Larsson, C.; Ruusuvuori, P.; Hartman, J.; Rantalainen, M. Predicting molecular phenotypes from histopathology images: A transcriptome-wide expression-morphology analysis in breast cancer. arXiv 2020, arXiv:2009.08917. [Google Scholar] [CrossRef]
- Chauhan, R.; Vinod, P.; Jawahar, C. Exploring Genetic-histologic Relationships in Breast Cancer. In Proceedings of the 2021 IEEE 18th International Symposium on Biomedical Imaging (ISBI), Nice, France, 13–16 April 2021; pp. 1187–1190. [Google Scholar]
- Sadhwani, A.; Chang, H.W.; Behrooz, A.; Brown, T.; Auvigne-Flament, I.; Patel, H.; Findlater, R.; Velez, V.; Tan, F.; Tekiela, K.; et al. Comparative analysis of machine learning approaches to classify tumor mutation burden in lung adenocarcinoma using histopathology images. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Diao, J.A.; Chui, W.F.; Wang, J.K.; Mitchell, R.N.; Rao, S.K.; Resnick, M.B.; Lahiri, A.; Maheshwari, C.; Glass, B.; Mountain, V.; et al. Dense, high-resolution mapping of cells and tissues from pathology images for the interpretable prediction of molecular phenotypes in cancer. bioRxiv 2020. [Google Scholar]
- AlGhamdiă, H.M.; Koohbanani, N.A.; Rajpoot, N.; Raza, S.E.A. A Novel Cell Map Representation for Weakly Supervised Prediction of ER & PR Status from H&E WSIs. In Proceedings of the MICCAI Workshop on Computational Pathology, PMLR, Strasbourg, France, 27 September 2021; pp. 10–19. [Google Scholar]
- Lu, W.; Graham, S.; Bilal, M.; Rajpoot, N.; Minhas, F. Capturing cellular topology in multi-gigapixel pathology images. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops, Seattle, WA, USA, 14–19 June 2020; pp. 260–261. [Google Scholar]
- Lu, W.; Toss, M.; Dawood, M.; Rakha, E.; Rajpoot, N.; Minhas, F. SlideGraph+: Whole Slide Image Level Graphs to Predict HER2 Status in Breast Cancer. Med. Image Anal. 2022, 80, 102486. [Google Scholar] [CrossRef]
- Gamble, P.; Jaroensri, R.; Wang, H.; Tan, F.; Moran, M.; Brown, T.; Flament-Auvigne, I.; Rakha, E.A.; Toss, M.; Dabbs, D.J.; et al. Determining breast cancer biomarker status and associated morphological features using deep learning. Commun. Med. 2021, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; Zheng, A.; Zhu, X.; Liu, S.; Hu, M.; Luo, Q.; Liao, H.; Liu, M.; He, Y.; et al. Predict Ki-67 positive cells in H&E-stained images using deep learning independently from IHC-stained images. Front. Mol. Biosci. 2020, 7, 183. [Google Scholar] [PubMed]
- Su, A.; Lee, H.; Tan, X.; Suarez, C.J.; Andor, N.; Nguyen, Q.; Ji, H.P. A deep learning model for molecular label transfer that enables cancer cell identification from histopathology images. NPJ Precis. Oncol. 2022, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wharton, K.A., Jr.; Wood, D.; Manesse, M.; Maclean, K.H.; Leiss, F.; Zuraw, A. Tissue multiplex analyte detection in anatomic pathology–pathways to clinical implementation. Front. Mol. Biosci. 2021, 8, 719. [Google Scholar] [CrossRef] [PubMed]
- Shamai, G.; Binenbaum, Y.; Slossberg, R.; Duek, I.; Gil, Z.; Kimmel, R. Artificial intelligence algorithms to assess hormonal status from tissue microarrays in patients with breast cancer. JAMA Netw. Open 2019, 2, e197700. [Google Scholar] [CrossRef] [Green Version]
- Muti, H.S.; Heij, L.R.; Keller, G.; Kohlruss, M.; Langer, R.; Dislich, B.; Cheong, J.H.; Kim, Y.W.; Kim, H.; Kook, M.C.; et al. Development and validation of deep learning classifiers to detect Epstein-Barr virus and microsatellite instability status in gastric cancer: A retrospective multicentre cohort study. Lancet Digit. Health 2021, 3, e654–e664. [Google Scholar] [CrossRef]
- Schrammen, P.L.; Ghaffari Laleh, N.; Echle, A.; Truhn, D.; Schulz, V.; Brinker, T.J.; Brenner, H.; Chang-Claude, J.; Alwers, E.; Brobeil, A.; et al. Weakly supervised annotation-free cancer detection and prediction of genotype in routine histopathology. J. Pathol. 2022, 256, 50–60. [Google Scholar] [CrossRef]
- Echle, A.; Laleh, N.G.; Quirke, P.; Grabsch, H.; Muti, H.; Saldanha, O.; Brockmoeller, S.; van den Brandt, P.; Hutchins, G.; Richman, S.; et al. Artificial intelligence for detection of microsatellite instability in colorectal cancer: A multicentric analysis of a pre-screening tool for clinical application. ESMO Open 2022, 7, 100400. [Google Scholar] [CrossRef]
- Wang, X.; Zou, C.; Zhang, Y.; Li, X.; Wang, C.; Ke, F.; Chen, J.; Wang, W.; Wang, D.; Xu, X.; et al. Prediction of BRCA gene mutation in breast cancer based on deep learning and histopathology images. Front. Genet. 2021, 12, 1147. [Google Scholar] [CrossRef]
- Schömig-Markiefka, B.; Pryalukhin, A.; Hulla, W.; Bychkov, A.; Fukuoka, J.; Madabhushi, A.; Achter, V.; Nieroda, L.; Büttner, R.; Quaas, A.; et al. Quality control stress test for deep learning-based diagnostic model in digital pathology. Mod. Pathol. 2021, 34, 2098–2108. [Google Scholar] [CrossRef]
- Ozyoruk, K.B.; Can, S.; Gokceler, G.I.; Basak, K.; Demir, D.; Serin, G.; Hacisalihoglu, U.P.; Darbaz, B.; Lu, M.Y.; Chen, T.Y.; et al. Deep Learning-based Frozen Section to FFPE Translation. arXiv 2021, arXiv:2107.11786. [Google Scholar]
- Seegerer, P.; Binder, A.; Saitenmacher, R.; Bockmayr, M.; Alber, M.; Jurmeister, P.; Klauschen, F.; Müller, K.R. Interpretable deep neural network to predict estrogen receptor status from haematoxylin-eosin images. In Artificial Intelligence and Machine Learning for Digital Pathology; Springer: Berlin/Heidelberg, Germany, 2020; pp. 16–37. [Google Scholar]
- Bychkov, D.; Linder, N.; Tiulpin, A.; Kücükel, H.; Lundin, M.; Nordling, S.; Sihto, H.; Isola, J.; Lehtimäki, T.; Kellokumpu-Lehtinen, P.L.; et al. Deep learning identifies morphological features in breast cancer predictive of cancer ERBB2 status and trastuzumab treatment efficacy. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Graham, S.; Vu, Q.D.; Raza, S.E.A.; Azam, A.; Tsang, Y.W.; Kwak, J.T.; Rajpoot, N. Hover-net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 2019, 58, 101563. [Google Scholar] [CrossRef]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef]
- Naik, N.; Madani, A.; Esteva, A.; Keskar, N.S.; Press, M.F.; Ruderman, D.; Agus, D.B.; Socher, R. Deep learning-enabled breast cancer hormonal receptor status determination from base-level H&E stains. Nat. Commun. 2020, 11, 1–8. [Google Scholar]
- Schmitt, M.; Maron, R.C.; Hekler, A.; Stenzinger, A.; Hauschild, A.; Weichenthal, M.; Tiemann, M.; Krahl, D.; Kutzner, H.; Utikal, J.S.; et al. Hidden variables in deep learning digital pathology and their potential to cause batch effects: Prediction model study. J. Med. Internet Res. 2021, 23, e23436. [Google Scholar] [CrossRef]
- Javed, S.A.; Juyal, D.; Shanis, Z.; Chakraborty, S.; Pokkalla, H.; Prakash, A. Rethinking Machine Learning Model Evaluation in Pathology. arXiv 2022, arXiv:2204.05205. [Google Scholar]
- Yamashita, R.; Long, J.; Banda, S.; Shen, J.; Rubin, D.L. Learning domain-agnostic visual representation for computational pathology using medically-irrelevant style transfer augmentation. IEEE Trans. Med. Imaging 2021, 40, 3945–3954. [Google Scholar] [CrossRef]
- Howard, F.M.; Dolezal, J.; Kochanny, S.; Schulte, J.; Chen, H.; Heij, L.; Huo, D.; Nanda, R.; Olopade, O.I.; Kather, J.N.; et al. The impact of digital histopathology batch effect on deep learning model accuracy and bias. BioRxiv 2020. [Google Scholar]
- Dehkharghanian, T.; Bidgoli, A.A.; Riasatian, A.; Mazaheri, P.; Campbell, C.J.; Pantanowitz, L.; Tizhoosh, H.; Rahnamayan, S. Biased Data, Biased AI: Deep Networks Predict the Acquisition Site of TCGA Images. Res. Sq. 2021. Europe PMC. Available online: https://www.researchsquare.com/article/rs-943804/v1 (accessed on 4 December 2022).
- Bustos, A.; Payá, A.; Torrubia, A.; Jover, R.; Llor, X.; Bessa, X.; Castells, A.; Carracedo, Á.; Alenda, C. XDEEP-MSI: Explainable Bias-Rejecting Microsatellite Instability Deep Learning System in Colorectal Cancer. Biomolecules 2021, 11, 1786. [Google Scholar] [CrossRef] [PubMed]
- Wiles, O.; Gowal, S.; Stimberg, F.; Alvise-Rebuffi, S.; Ktena, I.; Dvijotham, K.; Cemgil, T. A fine-grained analysis on distribution shift. arXiv 2021, arXiv:2110.11328. [Google Scholar]
- Oakden-Rayner, L.; Dunnmon, J.; Carneiro, G.; Ré, C. Hidden stratification causes clinically meaningful failures in machine learning for medical imaging. In Proceedings of the ACM Conference on Health Inference, and Learning, Toronto, ON, Canada, 2–4 April 2020; pp. 151–159. [Google Scholar]
- Galstyan, T.; Harutyunyan, H.; Khachatrian, H.; Steeg, G.V.; Galstyan, A. Failure Modes of Domain Generalization Algorithms. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Vancouver, BC, Canada, 19–20 June 2022; pp. 19077–19086. [Google Scholar]
- Ektefaie, Y.; Yuan, W.; Dillon, D.A.; Lin, N.U.; Golden, J.A.; Kohane, I.S.; Yu, K.H. Integrative multiomics-histopathology analysis for breast cancer classification. NPJ Breast Cancer 2021, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lazard, T.; Bataillon, G.; Naylor, P.; Popova, T.; Bidard, F.C.; Stoppa-Lyonnet, D.; Stern, M.H.; Decencière, E.; Walter, T.; Salomon, A.V. Deep Learning identifies new morphological patterns of Homologous Recombination Deficiency in luminal breast cancers from whole slide images. bioRxiv 2021. [Google Scholar]
- Krause, J.; Grabsch, H.I.; Kloor, M.; Jendrusch, M.; Echle, A.; Buelow, R.D.; Boor, P.; Luedde, T.; Brinker, T.J.; Trautwein, C.; et al. Deep learning detects genetic alterations in cancer histology generated by adversarial networks. J. Pathol. 2021, 254, 70–79. [Google Scholar] [CrossRef]
- Andreux, M.; Terrail, J.O.d.; Beguier, C.; Tramel, E.W. Siloed federated learning for multi-centric histopathology datasets. In Domain Adaptation and Representation Transfer, and Distributed and Collaborative Learning; Springer: Berlin/Heidelberg, Germany, 2020; pp. 129–139. [Google Scholar]
- Saldanha, O.L.; Quirke, P.; West, N.P.; James, J.A.; Loughrey, M.B.; Grabsch, H.I.; Salto-Tellez, M.; Alwers, E.; Cifci, D.; Ghaffari Laleh, N.; et al. Swarm learning for decentralized artificial intelligence in cancer histopathology. Nat. Med. 2022, 28, 1–8. [Google Scholar] [CrossRef]
- Jaber, M.I.; Song, B.; Taylor, C.; Vaske, C.J.; Benz, S.C.; Rabizadeh, S.; Soon-Shiong, P.; Szeto, C.W. A deep learning image-based intrinsic molecular subtype classifier of breast tumors reveals tumor heterogeneity that may affect survival. Breast Cancer Res. 2020, 22, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Arslan, S.; Mehrotra, D.; Schmidt, J.; Geraldes, A.; Singhal, S.; Hense, J.; Li, X.; Bass, C.; Raharja-Liu, P. Large-scale systematic feasibility study on the pan-cancer predictability of multi-omic biomarkers from whole slide images with deep learning. bioRxiv 2022. [Google Scholar]
- Bodalal, Z.; Trebeschi, S.; Nguyen-Kim, T.D.L.; Schats, W.; Beets-Tan, R. Radiogenomics: Bridging imaging and genomics. Abdom. Radiol. 2019, 44, 1960–1984. [Google Scholar] [CrossRef] [Green Version]
- Korfiatis, P.; Erickson, B. Deep learning can see the unseeable: Predicting molecular markers from MRI of brain gliomas. Clin. Radiol. 2019, 74, 367–373. [Google Scholar] [CrossRef]
- Abdurixiti, M.; Nijiati, M.; Shen, R.; Ya, Q.; Abuduxiku, N.; Nijiati, M. Current progress and quality of radiomic studies for predicting EGFR mutation in patients with non-small cell lung cancer using PET/CT images: A systematic review. Br. J. Radiol. 2021, 94, 20201272. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, T.; Han, M. The application of radiomics in predicting gene mutations in cancer. Eur. Radiol. 2022, 32, 1–11. [Google Scholar] [CrossRef]
- Zhang, N.; Wu, J.; Yu, J.; Zhu, H.; Yang, M.; Li, R. Integrating Imaging, Histologic, and Genetic Features to Predict Tumor Mutation Burden of Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2020, 21, e151–e163. [Google Scholar] [CrossRef]
- Yin, Q.; Hung, S.C.; Rathmell, W.K.; Shen, L.; Wang, L.; Lin, W.; Fielding, J.R.; Khandani, A.H.; Woods, M.E.; Milowsky, M.I.; et al. Integrative radiomics expression predicts molecular subtypes of primary clear cell renal cell carcinoma. Clin. Radiol. 2018, 73, 782–791. [Google Scholar] [CrossRef]
- Chen, C.L.; Chen, C.C.; Yu, W.H.; Chen, S.H.; Chang, Y.C.; Hsu, T.I.; Hsiao, M.; Yeh, C.Y.; Chen, C.Y. An annotation-free whole-slide training approach to pathological classification of lung cancer types using deep learning. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Pinckaers, H.; Van Ginneken, B.; Litjens, G. Streaming convolutional neural networks for end-to-end learning with multi-megapixel images. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 44, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Pinckaers, H.; Bulten, W.; van der Laak, J.; Litjens, G. Detection of prostate cancer in whole-slide images through end-to-end training with image-level labels. IEEE Trans. Med. Imaging 2021, 40, 1817–1826. [Google Scholar] [CrossRef]
- Hou, L.; Cheng, Y.; Shazeer, N.; Parmar, N.; Li, Y.; Korfiatis, P.; Drucker, T.M.; Blezek, D.J.; Song, X. High resolution medical image analysis with spatial partitioning. arXiv 2019, arXiv:1909.03108. [Google Scholar]
- Shao, Z.; Bian, H.; Chen, Y.; Wang, Y.; Zhang, J.; Ji, X.; Zhang, Y. Transmil: Transformer based correlated multiple instance learning for whole slide image classification. Adv. Neural Inf. Process. Syst. 2021, 34, 2136–2147. [Google Scholar]
- Wang, X.; Yang, S.; Zhang, J.; Wang, M.; Zhang, J.; Huang, J.; Yang, W.; Han, X. Transpath: Transformer-based self-supervised learning for histopathological image classification. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention; Springer: Berlin/Heidelberg, Germany, 2021; pp. 186–195. [Google Scholar]
- Chen, R.J.; Lu, M.Y.; Weng, W.H.; Chen, T.Y.; Williamson, D.F.; Manz, T.; Shady, M.; Mahmood, F. Multimodal co-attention transformer for survival prediction in gigapixel whole slide images. In Proceedings of the IEEE/CVF International Conference on Computer Vision, Montreal, BC, Canada, 11–17 October 2021; pp. 4015–4025. [Google Scholar]
- Stegmüller, T.; Spahr, A.; Bozorgtabar, B.; Thiran, J.P. Scorenet: Learning non-uniform attention and augmentation for transformer-based histopathological image classification. arXiv 2022, arXiv:2202.07570. [Google Scholar]
- Kather, J.N. Histological Images for MSI vs. MSS Classification in Gastrointestinal Cancer, FFPE Samples; Zenodo: Meyrin, Switzerland, 2019. [Google Scholar] [CrossRef]
- Kather, J.N. Image Tiles of TCGA-CRC-DX Histological Whole Slide Images, Non-Normalized, Tumor Only; Zenodo: Meyrin, Switzerland, 2020. [Google Scholar] [CrossRef]
- Conde-Sousa, E.; Vale, J.; Feng, M.; Xu, K.; Wang, Y.; Della Mea, V.; La Barbera, D.; Montahaei, E.; Baghshah, M.S.; Turzynski, A.; et al. HEROHE Challenge: Assessing HER2 status in breast cancer without immunohistochemistry or in situ hybridization. arXiv 2021, arXiv:2111.04738. [Google Scholar] [CrossRef]
- Conde-Sousa, E.; Vale, J.; Feng, M.; Xu, K.; Wang, Y.; Della Mea, V.; La Barbera, D.; Montahaei, E.; Baghshah, M.; Turzynski, A.; et al. HEROHE Challenge: Predicting HER2 Status in Breast Cancer from Hematoxylin–Eosin Whole-Slide Imaging. J. Imaging 2022, 8, 213. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couture, H.D. Deep Learning-Based Prediction of Molecular Tumor Biomarkers from H&E: A Practical Review. J. Pers. Med. 2022, 12, 2022. https://doi.org/10.3390/jpm12122022
Couture HD. Deep Learning-Based Prediction of Molecular Tumor Biomarkers from H&E: A Practical Review. Journal of Personalized Medicine. 2022; 12(12):2022. https://doi.org/10.3390/jpm12122022
Chicago/Turabian StyleCouture, Heather D. 2022. "Deep Learning-Based Prediction of Molecular Tumor Biomarkers from H&E: A Practical Review" Journal of Personalized Medicine 12, no. 12: 2022. https://doi.org/10.3390/jpm12122022
APA StyleCouture, H. D. (2022). Deep Learning-Based Prediction of Molecular Tumor Biomarkers from H&E: A Practical Review. Journal of Personalized Medicine, 12(12), 2022. https://doi.org/10.3390/jpm12122022