

IGFBP2 Drives Regulatory T Cell Differentiation through STAT3/IDO Signaling Pathway in Pancreatic Cancer

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. Immunohistochemical Analysis, Flow Cytometry and Enzyme-Linked Immunosorbent Assay
2.3. Patient-Derived Xenograft Cells and Cell Culture
2.4. Orthotopic PDAC Mouse Model
2.5. Lentivirus, Plasmids, and siRNAs
2.6. T Cell Studies
2.6.1. Treg Induction and T Cell Suppression
2.6.2. T Cell Apoptosis Assay
2.6.3. Cytotoxicity Assay
2.6.4. T Cell Proliferation and Cell Cycle Analysis
2.7. qRT-PCR
2.8. Immunoblot
2.9. Bioinformatic Analysis
2.10. Statistical Analysis
3. Results
3.1. IGFBP2 Overexpression Correlates with the Infiltration of Tregs in the Tumor Microenvironment and Tumor Progression in Clinical PDAC
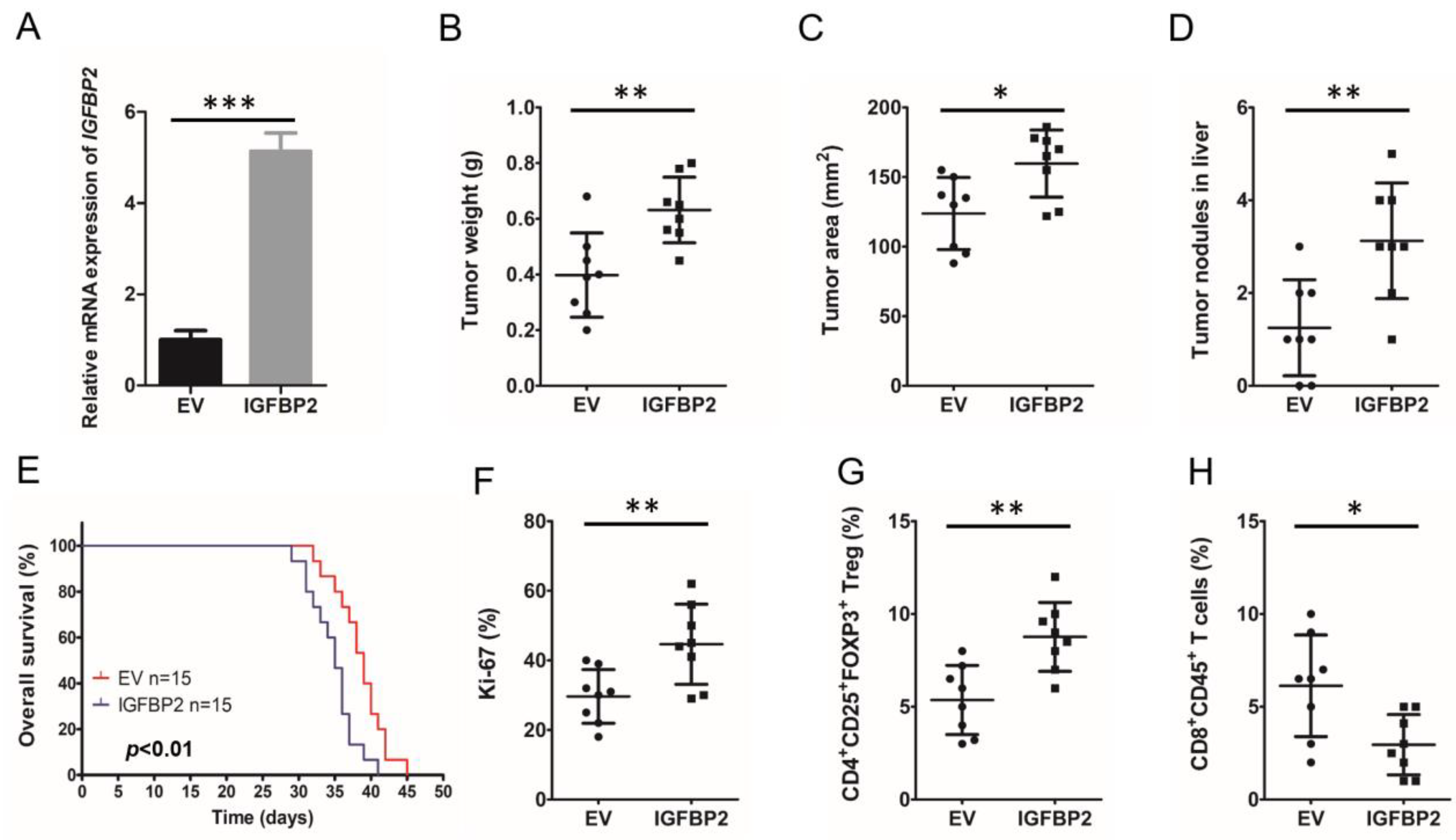
3.2. IGFBP2 Increases the Infiltration of Tregs in the TME and Promotes Disease Progression in Mouse PDAC
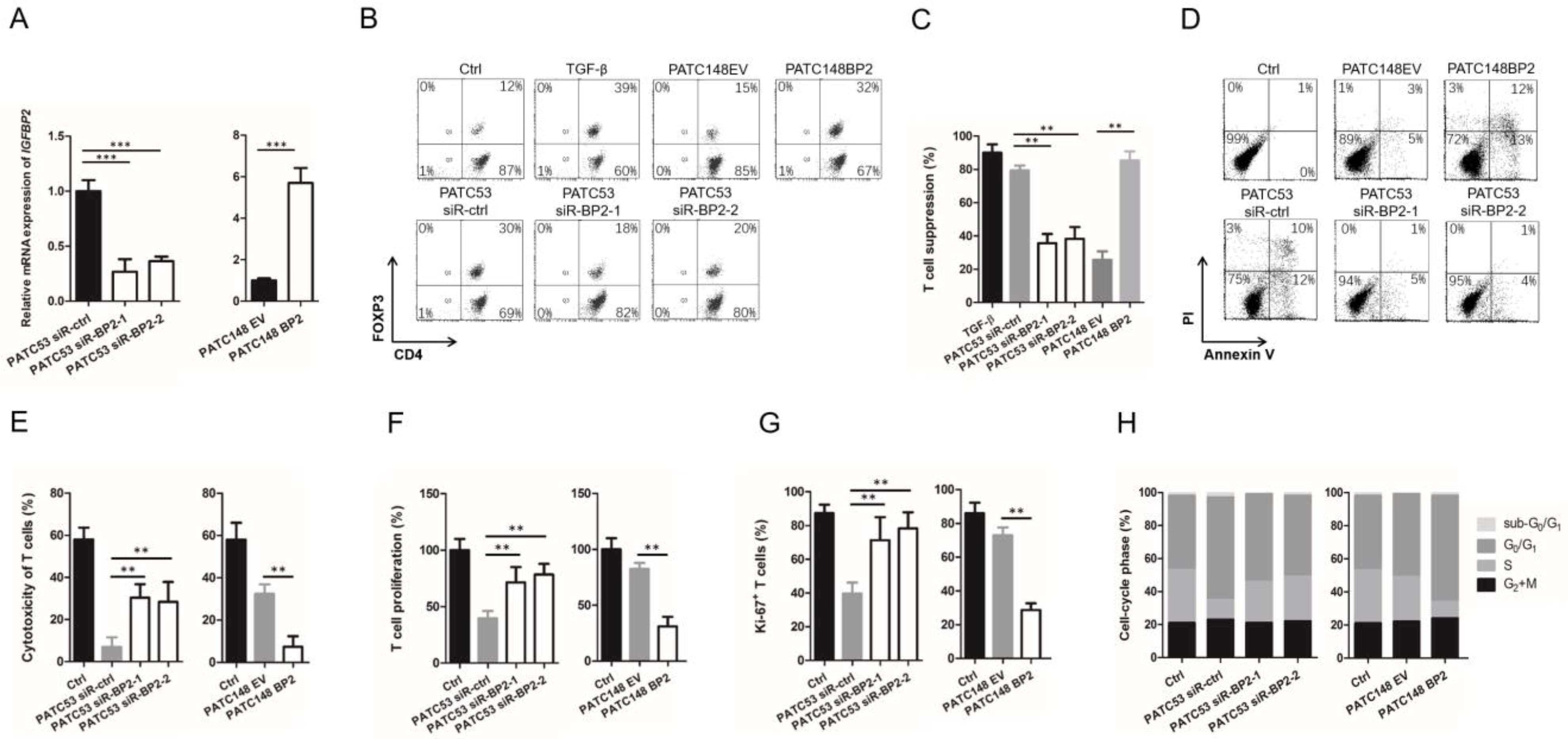
3.3. IGFBP2 Modifies T Cell Differentiation to an Immunosuppressive Phenotype
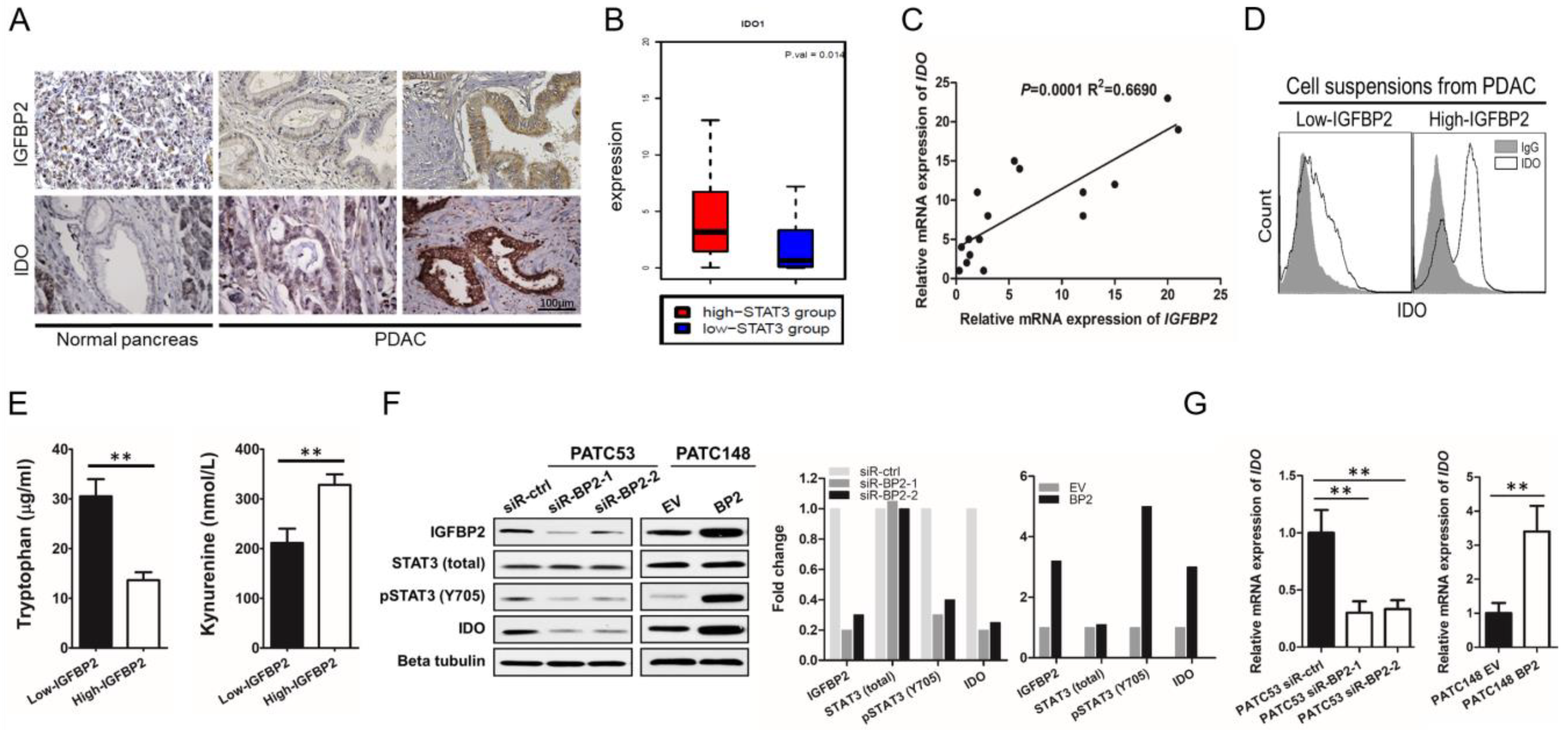
3.4. IGFBP2 Activates STAT3 and Induces IDO Expression in Human PDAC
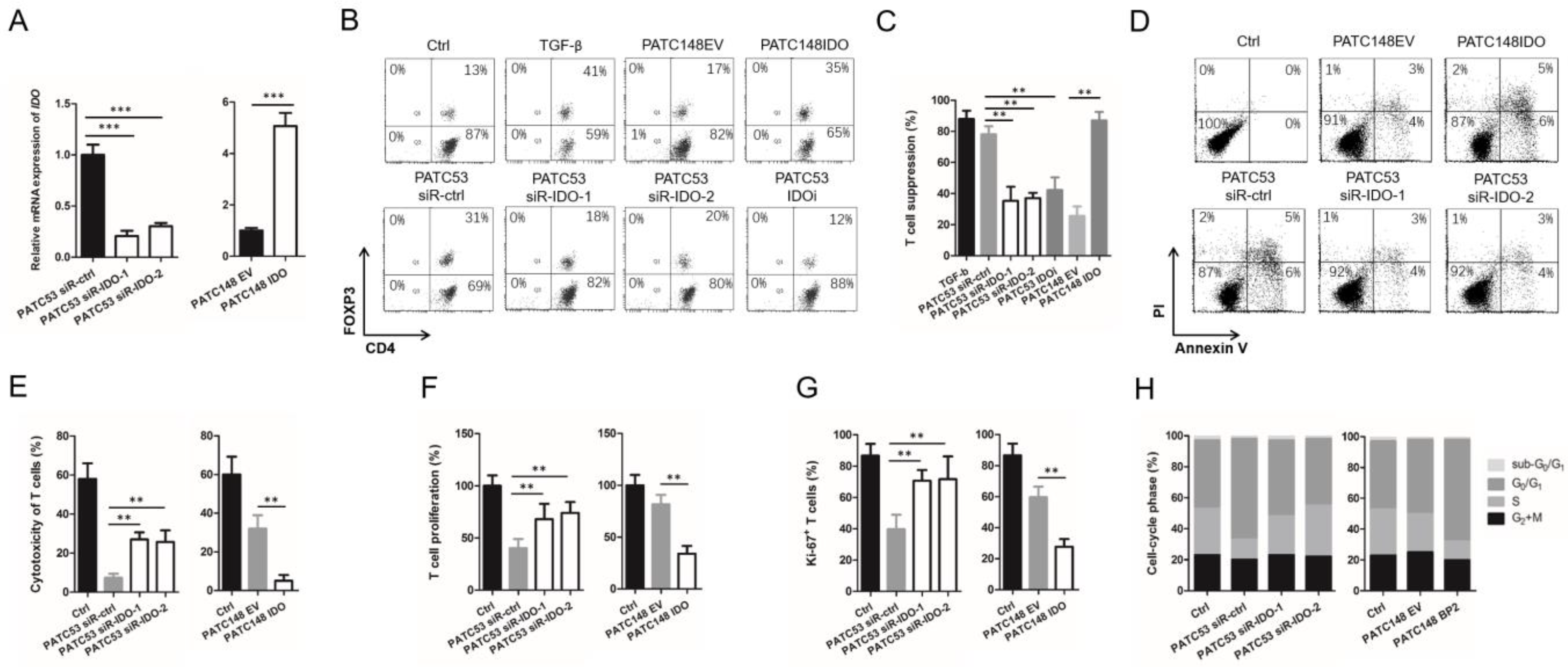
3.5. IGFBP2 Modifies T Cell Differentiation by Inducing IDO Expression in Human PDAC Cells
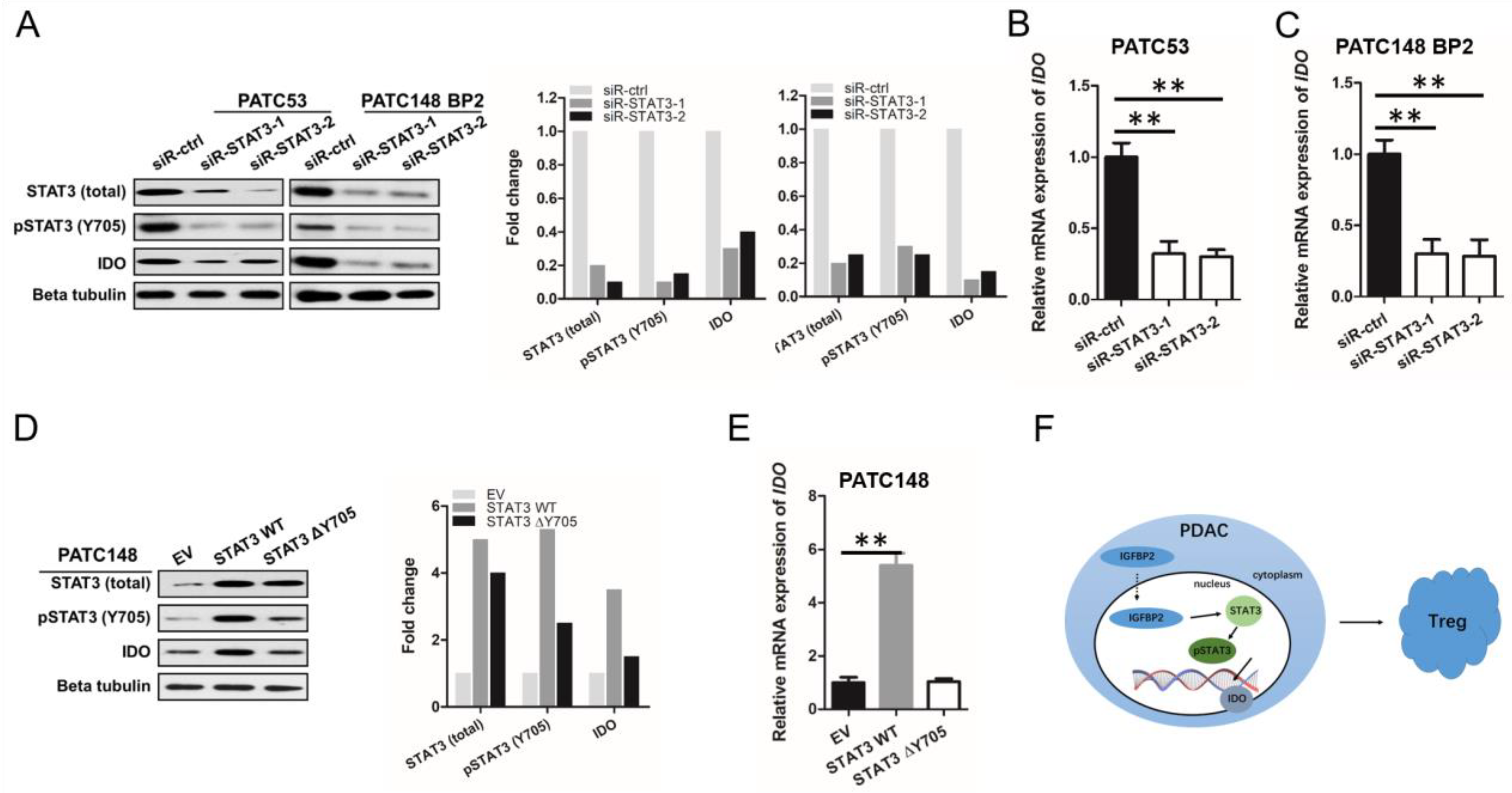
3.6. IGFBP2 Promotes IDO Production in Human PDAC via STAT3 Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.P. Pancreatic cancer epidemiology: Understanding the role of lifestyle and inherited risk factors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, H.; Lu, M.; Zhang, Y.; Lu, B.; You, L.; Zhang, T.; Dai, M.; Zhao, Y. Advances in the epidemiology of pancreatic cancer: Trends, risk factors, screening, and prognosis. Cancer Lett. 2021, 520, 1–11. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–1220. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.M.; Reiche, C.; Heiduk, M.; Tannert, A.; Meinecke, A.C.; Baier, S.; von Renesse, J.; Kahlert, C.; Distler, M.; Welsch, T.; et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene 2020, 39, 3102–3113. [Google Scholar] [CrossRef] [Green Version]
- Semaan, A.; Bernard, V.; Lee, J.J.; Wong, J.W.; Huang, J.; Swartzlander, D.B.; Stephens, B.M.; Monberg, M.E.; Weston, B.R.; Bhutani, M.S.; et al. Defining the Comprehensive Genomic Landscapes of Pancreatic Ductal Adenocarcinoma Using Real-World Endoscopic Aspiration Samples. Clin. Cancer Res. 2021, 27, 1082–1093. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [Green Version]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e13. [Google Scholar] [CrossRef] [Green Version]
- Peran, I.; Dakshanamurthy, S.; McCoy, M.D.; Mavropoulos, A.; Allo, B.; Sebastian, A.; Hum, N.R.; Sprague, S.C.; Martin, K.A.; Pishvaian, M.J.; et al. Cadherin 11 Promotes Immunosuppression and Extracellular Matrix Deposition to Support Growth of Pancreatic Tumors and Resistance to Gemcitabine in Mice. Gastroenterology 2021, 160, 1359–1372.e13. [Google Scholar] [CrossRef]
- Väyrynen, S.A.; Zhang, J.; Yuan, C.; Väyrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef]
- Takahashi, R.; Macchini, M.; Sunagawa, M.; Jiang, Z.; Tanaka, T.; Valenti, G.; Renz, B.W.; White, R.A.; Hayakawa, Y.; Westphalen, C.B.; et al. Interleukin-1β-induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte-mediated immune suppression. Gut 2021, 70, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673.e7. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Song, Q.; Liu, L.; Forbes, E.; Tian, W.; Zhang, Z.; Kang, Y.; Wang, H.; Fleming, J.B.; et al. IGFBP2 promotes tumor progression by inducing alternative polarization of macrophages in pancreatic ductal adenocarcinoma through the STAT3 pathway. Cancer Lett. 2021, 500, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Sun, Y.; Zhang, X.; Hu, L.; Liu, Y.; Chua, C.Y.; Phillips, L.M.; Ren, H.; Fleming, J.B.; Wang, H.; et al. IGFBP2 Activates the NF-κB Pathway to Drive Epithelial-Mesenchymal Transition and Invasive Character in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2016, 76, 6543–6554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef]
- Chen, H.; Bian, A.; Yang, L.F.; Yin, X.; Wang, J.; Ti, C.; Miao, Y.; Peng, S.; Xu, S.; Liu, M.; et al. Targeting STAT3 by a small molecule suppresses pancreatic cancer progression. Oncogene 2021, 40, 1440–1457. [Google Scholar] [CrossRef]
- Oweida, A.J.; Darragh, L.; Phan, A.; Binder, D.; Bhatia, S.; Mueller, A.; Court, B.V.; Milner, D.; Raben, D.; Woessner, R.; et al. STAT3 Modulation of Regulatory T Cells in Response to Radiation Therapy in Head and Neck Cancer. J. Natl. Cancer Inst. 2019, 111, 1339–1349. [Google Scholar] [CrossRef]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11, 1185. [Google Scholar] [CrossRef]
- Sun, Y.; Yokoi, K.; Li, H.; Gao, J.; Hu, L.; Liu, B.; Chen, K.; Hamilton, S.R.; Fan, D.; Sun, B.; et al. NGAL expression is elevated in both colorectal adenoma-carcinoma sequence and cancer progression and enhances tumorigenesis in xenograft mouse models. Clin. Cancer Res. 2011, 17, 4331–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Zhang, R.; Suzuki, R.; Li, S.Q.; Roife, D.; Truty, M.J.; Chatterjee, D.; Thomas, R.M.; Cardwell, J.; Wang, Y.; et al. Two-dimensional culture of human pancreatic adenocarcinoma cells results in an irreversible transition from epithelial to mesenchymal phenotype. Lab. Investig. 2015, 95, 207–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Zillmann, U.; Sprengel, R. Alternative Anesthesia of Neonatal Mice for Global rAAV Delivery in the Brain with Non-detectable Behavioral Interference in Adults. Front. Behav. Neurosci. 2020, 14, 115. [Google Scholar] [CrossRef]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.; Glennie, M.J.; et al. Committee of the National Cancer Research Institute. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadpour, H.; MacDonald, C.R.; Qiao, G.; Chen, M.; Dong, B.; Hylander, B.L.; McCarthy, P.L.; Abrams, S.I.; Repasky, E.A. β2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J. Clin. Investig. 2019, 129, 5537–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Yao, S.; Hu, Y.; Feng, Y.; Li, M.; Bian, Z.; Zhang, J.; Qin, Y.; Qi, X.; Zhou, L.; et al. The Immune-microenvironment Confers Chemoresistance of Colorectal Cancer through Macrophage-Derived IL6. Clin. Cancer Res. 2017, 23, 7375–7387. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lv, J.; Liu, J.; Liang, X.; Jin, X.; Xie, J.; Zhang, L.; Chen, D.; Fiskesund, R.; Tang, K.; et al. STAT3/p53 pathway activation disrupts IFN-β-induced dormancy in tumor-repopulating cells. J. Clin. Investig. 2018, 128, 1057–1073. [Google Scholar] [CrossRef] [Green Version]
- Alvisi, G.; Brummelman, J.; Puccio, S.; Mazza, E.M.; Tomada, E.P.; Losurdo, A.; Zanon, V.; Peano, C.; Colombo, F.S.; Scarpa, A.; et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Investig. 2020, 130, 3137–3150. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, C.; Zhao, G.; Zhang, X.; Hao, M.; Hassan, S.; Zhang, M.; Zheng, H.; Yang, D.; Liu, L.; et al. IGFBP2 regulates PD-L1 expression by activating the EGFR-STAT3 signaling pathway in malignant melanoma. Cancer Lett. 2020, 477, 19–30. [Google Scholar] [CrossRef]
- Sharma, M.D.; Pacholczyk, R.; Shi, H.; Berrong, Z.J.; Zakharia, Y.; Greco, A.; Chang, C.S.; Eathiraj, S.; Kennedy, E.; Cash, T.; et al. Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity 2021, 54, 2354–2371.e8. [Google Scholar] [CrossRef]
- Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Ellebaek, E.; Donia, M.; Holmstroem, R.B.; Klausen, T.W.; Madsen, C.O.; Ahmed, S.M.; Weis-Banke, S.E.; et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat. Med. 2021, 27, 2212–2223. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wu, R.; Li, K.; Yuan, W.; Zeng, C.; Zhang, Y.; Wang, X.; Zhu, X.; Zhou, J.; Li, P.; et al. IDO Inhibition Facilitates Antitumor Immunity of Vγ9Vδ2 T Cells in Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 679517. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Chesik, D.; De Keyser, J.; Wilczak, N. Involvement of insulin-like growth factor binding protein-2 in activated microglia as assessed in post mortem human brain. Neurosci. Lett. 2004, 362, 14–16. [Google Scholar] [CrossRef]
- Elmlinger, M.W.; Deininger, M.H.; Schuett, B.S.; Meyermann, R.; Duffner, F.; Grote, E.H.; Ranke, M.B. In vivo expression of insulin-like growth factor-binding protein-2 in human gliomas increases with the tumor grade. Endocrinology 2001, 142, 1652–1658. [Google Scholar] [CrossRef]
- Liu, Y.; Song, C.; Shen, F.; Zhang, J.; Song, S.W. IGFBP2 promotes immunosuppression associated with its mesenchymal induction and FcγRIIB phosphorylation in glioblastoma. PLoS ONE 2019, 14, e0222999. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Chen, Q.; Cui, Y.; Dong, J.; Chen, M.; Wu, P.; Jiang, C. Immune heterogeneity and clinicopathologic characterization of IGFBP2 in 2447 glioma samples. Oncoimmunology 2018, 7, e1426516. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Zhang, Y.; Yang, T.; Chen, J.; Zhang, X.; Liang, X. IGFBP2 Drives Regulatory T Cell Differentiation through STAT3/IDO Signaling Pathway in Pancreatic Cancer. J. Pers. Med. 2022, 12, 2005. https://doi.org/10.3390/jpm12122005
Sun L, Zhang Y, Yang T, Chen J, Zhang X, Liang X. IGFBP2 Drives Regulatory T Cell Differentiation through STAT3/IDO Signaling Pathway in Pancreatic Cancer. Journal of Personalized Medicine. 2022; 12(12):2005. https://doi.org/10.3390/jpm12122005
Chicago/Turabian StyleSun, Longhao, Yang Zhang, Tiantian Yang, Junhang Chen, Xuebin Zhang, and Xiaoyu Liang. 2022. "IGFBP2 Drives Regulatory T Cell Differentiation through STAT3/IDO Signaling Pathway in Pancreatic Cancer" Journal of Personalized Medicine 12, no. 12: 2005. https://doi.org/10.3390/jpm12122005
APA StyleSun, L., Zhang, Y., Yang, T., Chen, J., Zhang, X., & Liang, X. (2022). IGFBP2 Drives Regulatory T Cell Differentiation through STAT3/IDO Signaling Pathway in Pancreatic Cancer. Journal of Personalized Medicine, 12(12), 2005. https://doi.org/10.3390/jpm12122005