
Towards Precision Oncology: The Role of Smoothened and Its Variants in Cancer


Abstract
1. Introduction
2. The Structure of Smoothened
2.1. Domain Architecture and Binding Pockets
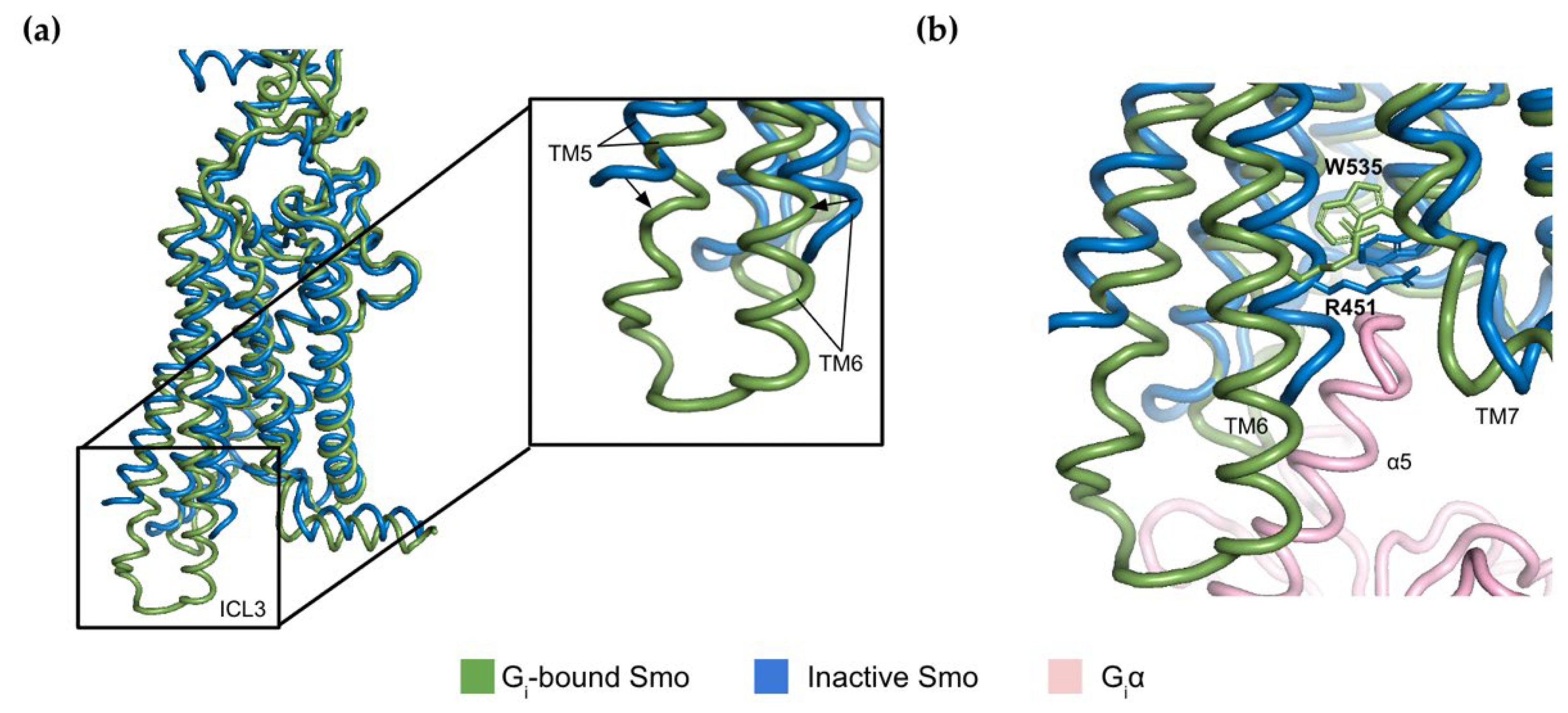
2.2. The Molecular Switch Mechanism of Activation
3. Hedgehog Signalling and Regulation of Smoothened
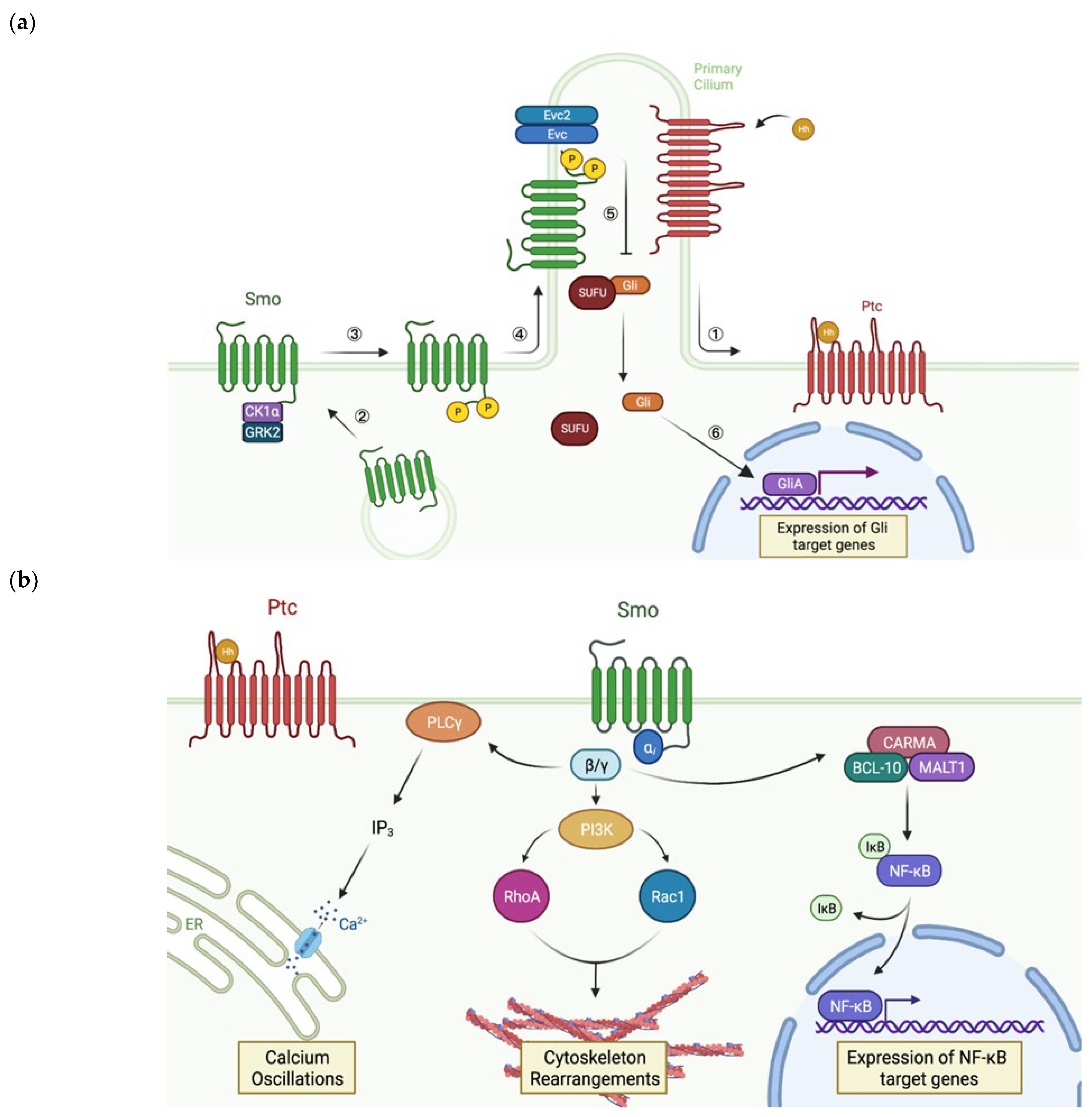
3.1. Smoothened and the Hedgehog Pathway
3.2. Regulation of Smoothened Activity
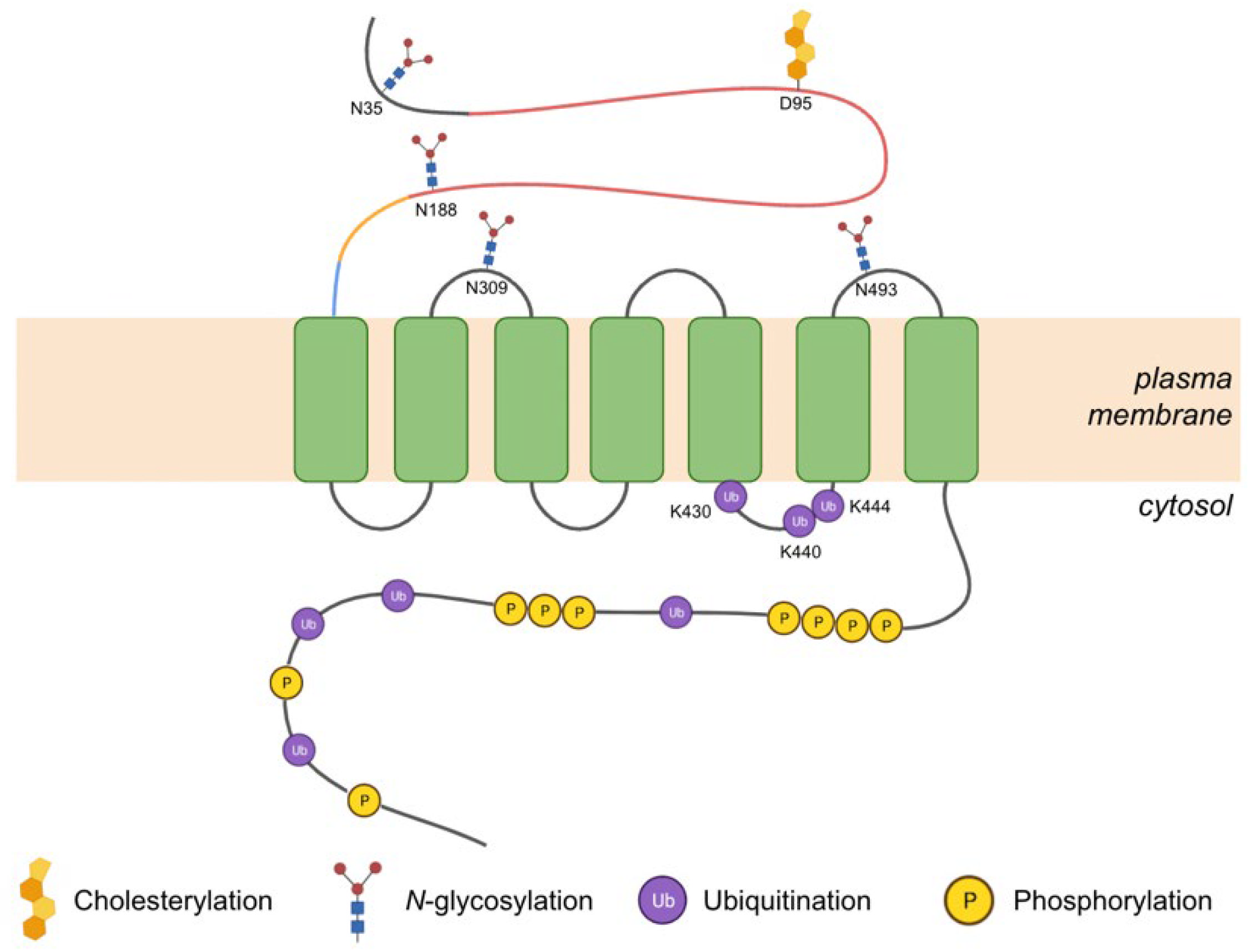
3.2.1. Post-Translational Modifications
3.2.2. Small Molecules
4. Targeting Smoothened in Cancer
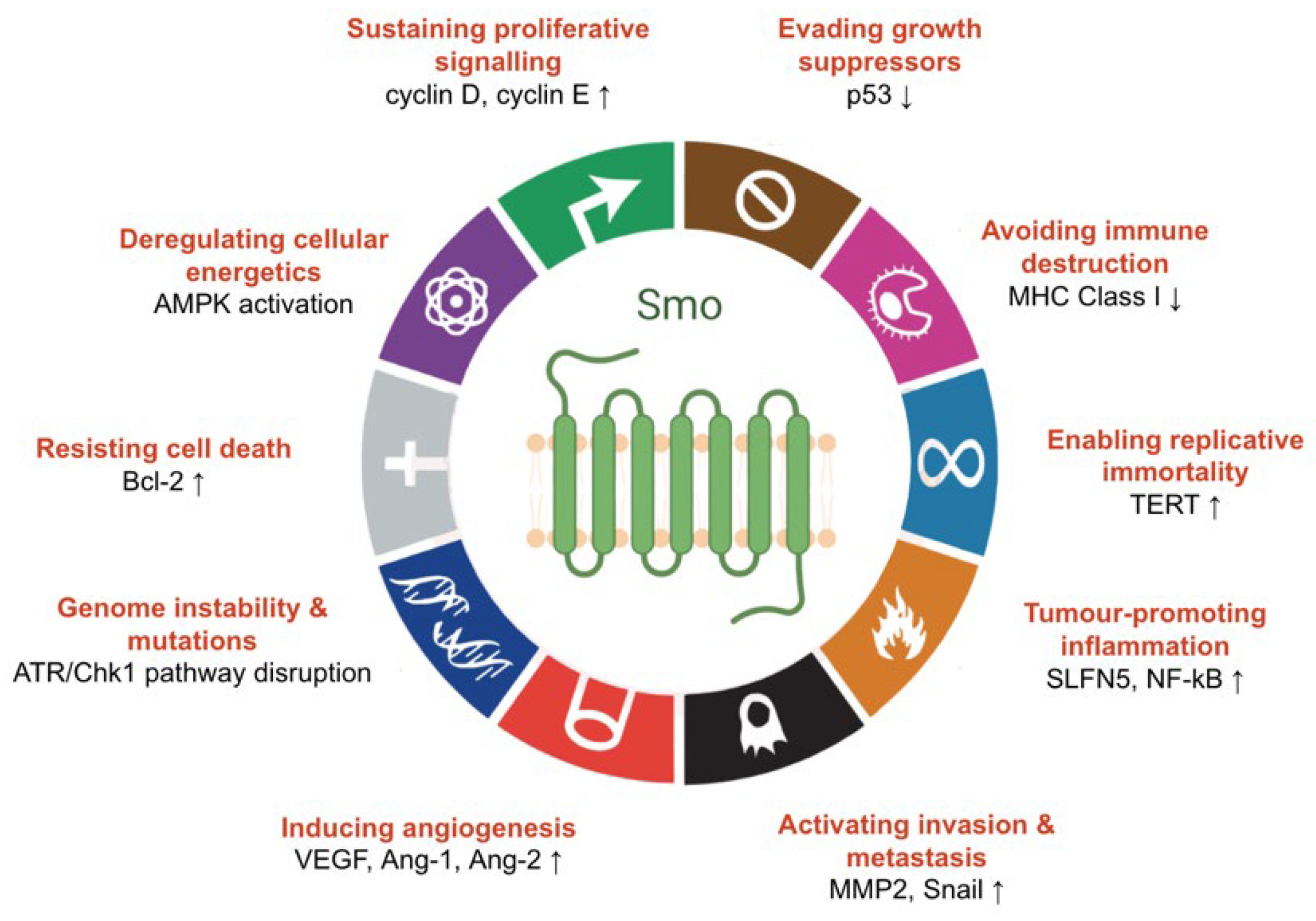
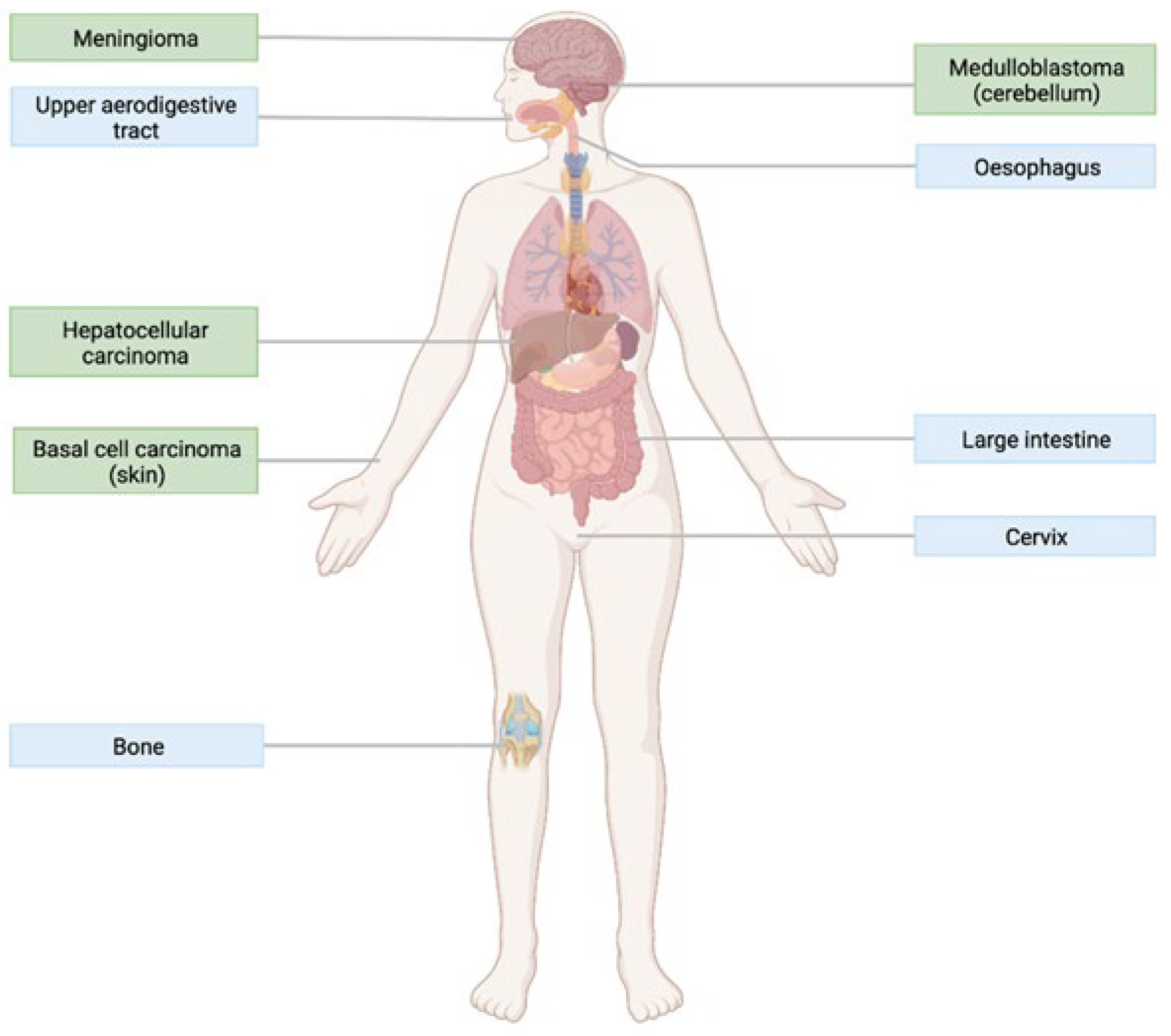
4.1. The Role of Hedgehog Signalling in Cancer

4.2. Smoothened as a Therapeutic Target
5. Smoothened Variants and Their Impact on Cancer Therapy
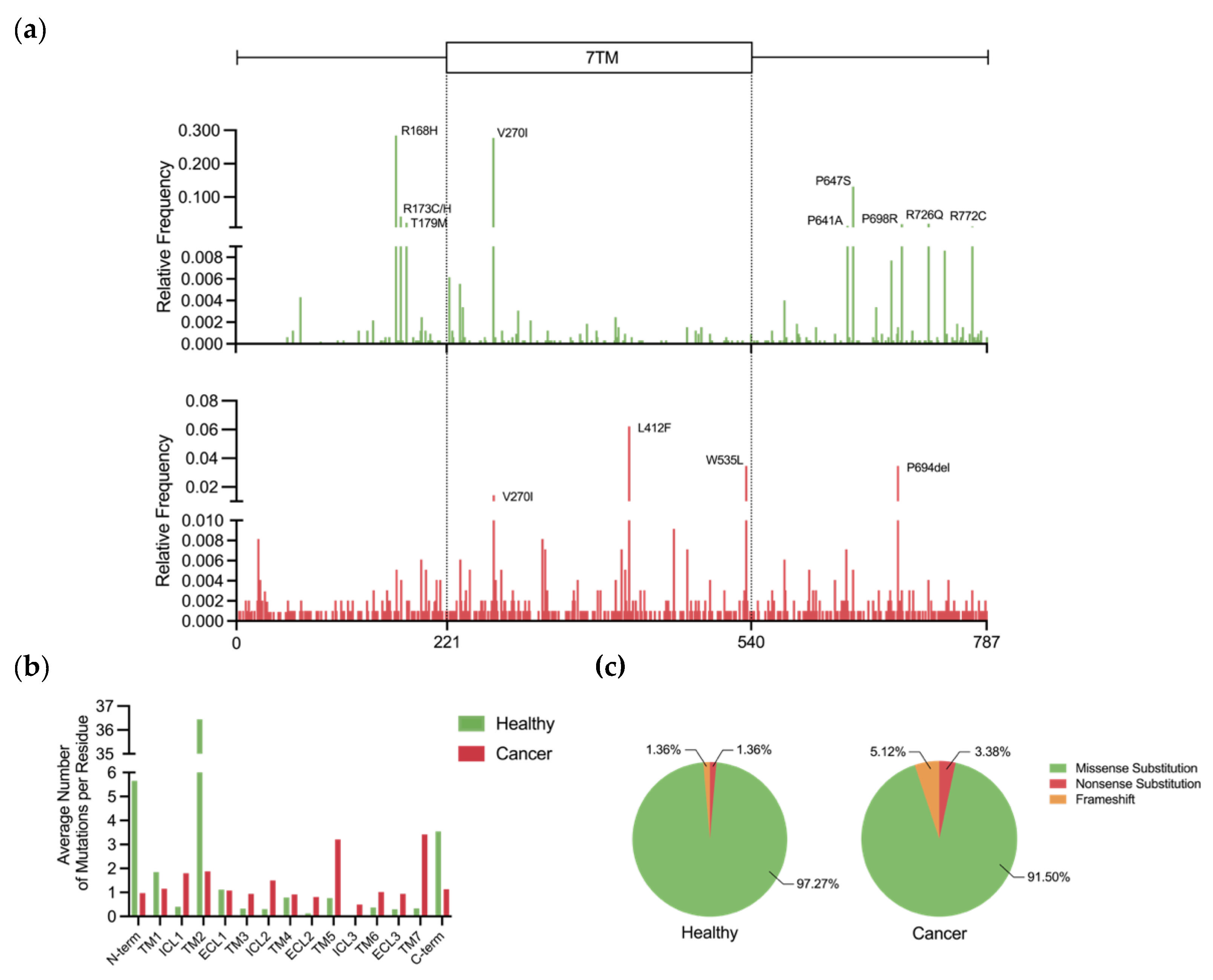
5.1. Prevalence and Incidence of Genetic Variation
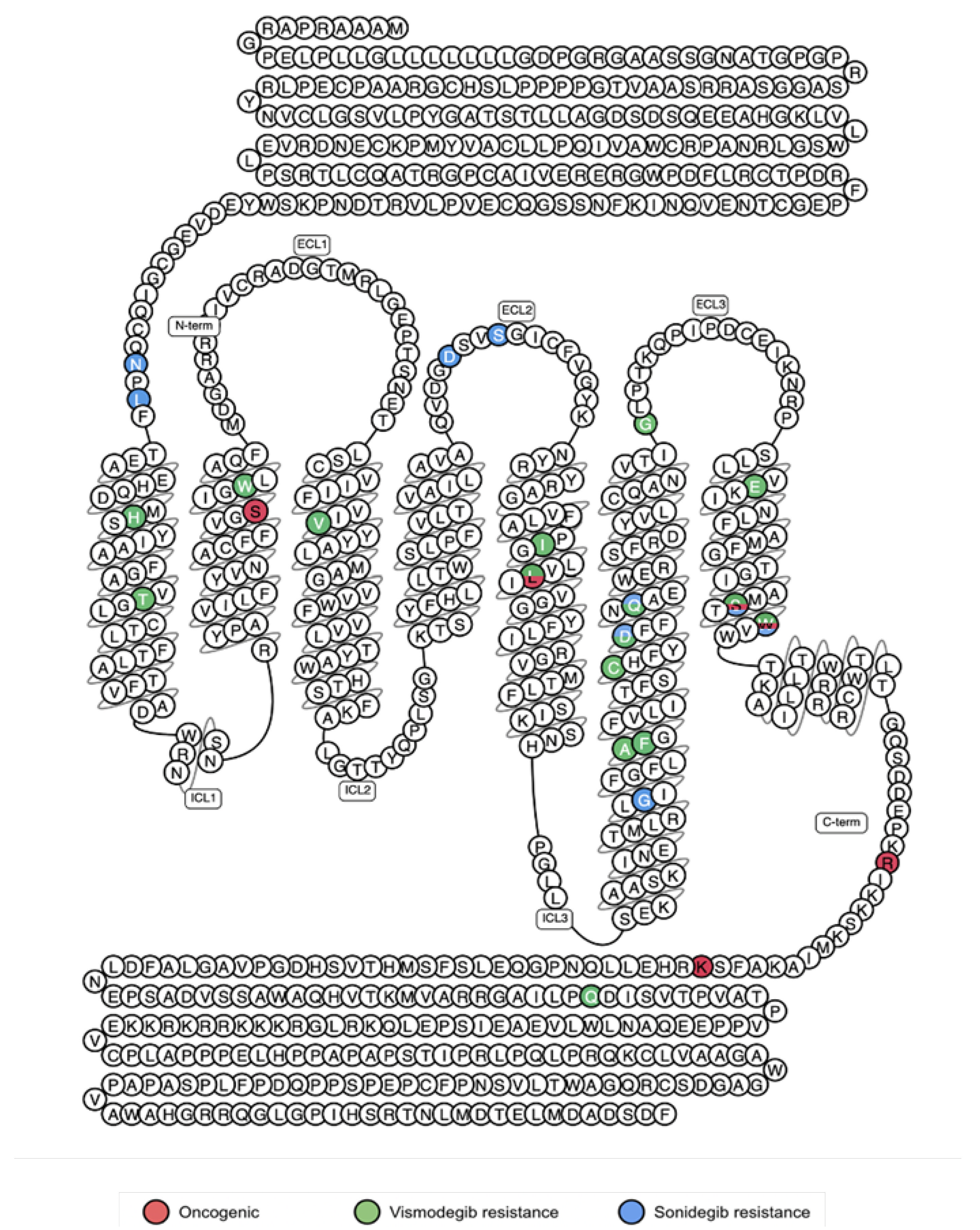
5.2. Activating Mutations
5.3. Resistance Mutations
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G Protein-Coupled Receptors: Structure- and Function-Based Drug Discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Ruat, M.; Hoch, L.; Faure, H.; Rognan, D. Targeting of Smoothened for Therapeutic Gain. Trends Pharmacol. Sci. 2014, 35, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.A.; Zhang, J.; Ho, C.; Cheung, S.-T.; Fan, S.-T.; Chen, X. Hedgehog Signaling in Human Hepatocellular Carcinoma. Cancer Biol. Ther. 2006, 5, 111–117. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.; Yajnik, V.; et al. Hedgehog Is an Early and Late Mediator of Pancreatic Cancer Tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog Signaling Pathway Is a New Therapeutic Target for Patients with Breast Cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef]
- Otsuka, A.; Levesque, M.P.; Dummer, R.; Kabashima, K. Hedgehog Signaling in Basal Cell Carcinoma. J. Dermatol. Sci. 2015, 78, 95–100. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Li, Y.-X.; Jayaraman, A.; Kannangai, R.; Qi, Y.; Vivekanandan, P.; Ludlow, J.W.; Owzar, K.; Chen, W.; Torbenson, M.S.; et al. Dysregulation of the Hedgehog Pathway in Human Hepatocarcinogenesis. Carcinogenesis 2006, 27, 748–757. [Google Scholar] [CrossRef]
- Walter, K.; Omura, N.; Hong, S.-M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of Smoothened Activates the Sonic Hedgehog Signaling Pathway in Pancreatic Cancer–Associated Fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef]
- Xie, J.; Murone, M.; Luoh, S.-M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.-W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened Mutations in Sporadic Basal-Cell Carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Khamaysi, Z.; Bochner, R.; Indelman, M.; Magal, L.; Avitan-Hersh, E.; Sarig, O.; Sprecher, E.; Bergman, R. Segmental Basal Cell Naevus Syndrome Caused by an Activating Mutation in Smoothened. Br. J. Dermatol. 2016, 175, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic Sequencing of Meningiomas Identifies Oncogenic SMO and AKT1 Mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the Genomic Complexity Underlying Medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.-S.; Sheen, I.-S.; Leu, C.-M.; Tseng, P.-H.; Chang, C.-F. The Role of Smoothened in Cancer. Int. J. Mol. Sci. 2020, 21, 6863. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR Sequence, Structure and Function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.G.; Bristow, M.R. The Emerging Pharmacogenomics of the Β-Adrenergic Receptors. Congest. Heart Fail. 2004, 10, 281–288. [Google Scholar] [CrossRef]
- Schulte, G.; Bryja, V. The Frizzled Family of Unconventional G-Protein-Coupled Receptors. Trends Pharmacol. Sci. 2007, 28, 518–525. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, F.; Wu, Y.; Yang, J.; Han, G.W.; Zhao, S.; Ishchenko, A.; Ye, L.; Lin, X.; Ding, K.; et al. Crystal Structure of a Multi-Domain Human Smoothened Receptor in Complex with a Super Stabilizing Ligand. Nat. Commun. 2017, 8, 15383. [Google Scholar] [CrossRef]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.; et al. The Tumour-Suppressor Gene Patched Encodes a Candidate Receptor for Sonic Hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef]
- Stark, D.R. Hedgehog Signalling: Pulling Apart Patched and Smoothened. Curr. Biol. 2002, 12, R437–R439. [Google Scholar] [CrossRef]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched Acts Catalytically to Suppress the Activity of Smoothened. Nature 2002, 418, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, I.; Liang, J.; Hedeen, D.; Roberts, K.J.; Zhang, Y.; Ha, B.; Latorraca, N.R.; Faust, B.; Dror, R.O.; Beachy, P.A.; et al. Smoothened Stimulation by Membrane Sterols Drives Hedgehog Pathway Activity. Nature 2019, 571, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Friedberg, L.; De Bose-Boyd, R.; Long, T.; Li, X. Sterols in an Intramolecular Channel of Smoothened Mediate Hedgehog Signaling. Nat. Chem. Biol. 2020, 16, 1368–1375. [Google Scholar] [CrossRef]
- Qi, X.; Liu, H.; Thompson, B.; McDonald, J.; Zhang, C.; Li, X. Cryo-EM Structure of Oxysterol-Bound Human Smoothened Coupled to a Heterotrimeric Gi. Nature 2019, 571, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.F.X.; Sircar, R.; Miller, P.S.; Hedger, G.; Luchetti, G.; Nachtergaele, S.; Tully, M.D.; Mydock-McGrane, L.; Covey, D.F.; Rambo, R.P.; et al. Structural Basis of Smoothened Regulation by Its Extracellular Domains. Nature 2016, 535, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.e14. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.-P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the Human Smoothened Receptor Bound to an Antitumour Agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Evron, T.; Vardy, E.; Han, G.W.; Huang, X.-P.; Hufeisen, S.J.; Mangano, T.J.; Urban, D.J.; Katritch, V.; et al. Structural Basis for Smoothened Receptor Modulation and Chemoresistance to Anticancer Drugs. Nat. Commun. 2014, 5, 4355. [Google Scholar] [CrossRef]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.H.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic Cubic Phase Injector Facilitates Membrane Protein Serial Femtosecond Crystallography. Nat. Commun. 2014, 5, 3309. [Google Scholar] [CrossRef]
- Wright, S.C.; Kozielewicz, P.; Kowalski-Jahn, M.; Petersen, J.; Bowin, C.-F.; Slodkowicz, G.; Marti-Solano, M.; Rodríguez, D.; Hot, B.; Okashah, N.; et al. A Conserved Molecular Switch in Class F Receptors Regulates Receptor Activation and Pathway Selection. Nat. Commun. 2019, 10, 667. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef] [PubMed]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog Signaling in Adult Organ Homeostasis and Repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.E.; Chiang, C. Hedgehog Secretion and Signal Transduction in Vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog Dependent Phosphorylation by CK1α and GRK2 Is Required for Ciliary Accumulation and Activation of Smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Chen, M.-H.; Chuang, P.-T. Smoothened Adopts Multiple Active and Inactive Conformations Capable of Trafficking to the Primary Cilium. PLoS ONE 2009, 4, e5182. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened Functions at the Primary Cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened Transduces Hedgehog Signal by Forming a Complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A Mechanism for Vertebrate Hedgehog Signaling: Recruitment to Cilia and Dissociation of SuFu–Gli Protein Complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef]
- Akhshi, T.; Trimble, W.S. A Non-Canonical Hedgehog Pathway Initiates Ciliogenesis and Autophagy. J. Cell Biol. 2021, 220, e202004179. [Google Scholar] [CrossRef]
- Belgacem, Y.H.; Borodinsky, L.N. Sonic Hedgehog Signaling Is Decoded by Calcium Spike Activity in the Developing Spinal Cord. Proc. Natl. Acad. Sci. USA 2011, 108, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Amann, S.; Bayer, M.; McGee, S.L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; et al. Hedgehog Partial Agonism Drives Warburg-like Metabolism in Muscle and Brown Fat. Cell 2012, 151, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi Proteins Link Hedgehog Signaling to Activation of Rho Small GTPases to Promote Fibroblast Migration. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, P.; Xiao, L.; Kazanietz, M.G.; Riobo, N.A. Hedgehog Proteins Activate Pro-Angiogenic Responses in Endothelial Cells through Non-Canonical Signaling Pathways. Cell Cycle 2010, 9, 570–579. [Google Scholar] [CrossRef]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.S.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway–Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with Resistance to Smoothened Antagonists by Inhibition of the PI3K Pathway in Medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef]
- Qu, C.; Liu, Y.; Kunkalla, K.; Singh, R.R.; Blonska, M.; Lin, X.; Agarwal, N.K.; Vega, F. Trimeric G Protein-CARMA1 Axis Links Smoothened, the Hedgehog Receptor Transducer, to NF-ΚB Activation in Diffuse Large B-Cell Lymphoma. Blood 2013, 121, 4718–4728. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Ma, G.; Li, S.; Han, Y.; Li, S.; Yue, T.; Wang, B.; Jiang, J. Regulation of Smoothened Trafficking and Hedgehog Signaling by the SUMO Pathway. Dev. Cell 2016, 39, 438–451. [Google Scholar] [CrossRef]
- Shi, D.; Lv, X.; Zhang, Z.; Yang, X.; Zhou, Z.; Zhang, L.; Zhao, Y. Smoothened Oligomerization/Higher Order Clustering in Lipid Rafts Is Essential for High Hedgehog Activity Transduction. J. Biol. Chem. 2013, 288, 12605–12614. [Google Scholar] [CrossRef]
- Okashah, N.; Wright, S.C.; Kawakami, K.; Mathiasen, S.; Zhou, J.; Lu, S.; Javitch, J.A.; Inoue, A.; Bouvier, M.; Lambert, N.A. Agonist-Induced Formation of Unproductive Receptor-G 12 Complexes. Proc. Natl. Acad. Sci. USA 2020, 117, 21723–21730. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ren, X.-R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; de Sauvage, F.; Lefkowitz, R.J. Activity-Dependent Internalization of Smoothened Mediated by ß-Arrestin 2 and GRK2. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Marada, S.; Navarro, G.; Truong, A.; Stewart, D.P.; Arensdorf, A.M.; Nachtergaele, S.; Angelats, E.; Opferman, J.T.; Rohatgi, R.; McCormick, P.J.; et al. Functional Divergence in the Role of N-Linked Glycosylation in Smoothened Signaling. PLoS Genet. 2015, 11, e1005473. [Google Scholar] [CrossRef]
- Wang, W.; Tian, Y.; Shi, X.; Ma, Q.; Xu, Y.; Yang, G.; Yi, W.; Shi, Y.; Zhou, N. N-glycosylation of the Human Neuropeptide QRFP Receptor (QRFPR) Is Essential for Ligand Binding and Receptor Activation. J. Neurochem. 2021, 158, 138–152. [Google Scholar] [CrossRef]
- Lee, S.-M.; Jeong, Y.; Simms, J.; Warner, M.L.; Poyner, D.R.; Chung, K.Y.; Pioszak, A.A. Calcitonin Receptor N-Glycosylation Enhances Peptide Hormone Affinity by Controlling Receptor Dynamics. J. Mol. Biol. 2020, 432, 1996–2014. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.B.; Stuck, M.W.; Lv, B.; Pazour, G.J. Ubiquitin Links Smoothened to Intraflagellar Transport to Regulate Hedgehog Signaling. J. Cell Biol. 2020, 219, e201912104. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Tang, J.-J.; Peng, C.; Wang, Y.; Fu, L.; Qiu, Z.-P.; Xiong, Y.; Yang, L.-F.; Cui, H.-W.; He, X.-L.; et al. Cholesterol Modification of Smoothened Is Required for Hedgehog Signaling. Mol. Cell 2017, 66, 154–162.e10. [Google Scholar] [CrossRef]
- Hu, A.; Zhang, J.-Z.; Wang, J.; Li, C.-C.; Yuan, M.; Deng, G.; Lin, Z.-C.; Qiu, Z.-P.; Liu, H.-Y.; Wang, X.-W.; et al. Cholesterylation of Smoothened Is a Calcium-Accelerated Autoreaction Involving an Intramolecular Ester Intermediate. Cell Res. 2022, 32, 288–301. [Google Scholar] [CrossRef]
- Nachtergaele, S.; Mydock, L.K.; Krishnan, K.; Rammohan, J.; Schlesinger, P.H.; Covey, D.F.; Rohatgi, R. Oxysterols Are Allosteric Activators of the Oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. [Google Scholar] [CrossRef]
- Nedelcu, D.; Liu, J.; Xu, Y.; Jao, C.; Salic, A. Oxysterol Binding to the Extracellular Domain of Smoothened in Hedgehog Signaling. Nat. Chem. Biol. 2013, 9, 557–564. [Google Scholar] [CrossRef]
- Myers, B.R.; Sever, N.; Chong, Y.C.; Kim, J.; Belani, J.D.; Rychnovsky, S.; Bazan, J.F.; Beachy, P.A. Hedgehog Pathway Modulation by Multiple Lipid Binding Sites on the Smoothened Effector of Signal Response. Dev. Cell 2013, 26, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The Teratogenic Veratrum Alkaloid Cyclopamine Inhibits Sonic Hedgehog Signal Transduction. Development 1998, 125, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zheng, S.; Wierbowski, B.M.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324.e16. [Google Scholar] [CrossRef]
- Tanaka, H.; Nakamura, M.; Kameda, C.; Kubo, M.; Sato, N.; Kuroki, S.; Tanaka, T.; Katano, M. The Hedgehog Signaling Pathway Plays an Essential Role in Maintaining the CD44+CD24-/low Subpopulation and the Side Population of Breast Cancer Cells. Anticancer Res. 2009, 29, 2147. [Google Scholar]
- Chen, X.; Cheng, Q.; She, M.; Wang, Q.; Huang, X.; Cao, L.; Fu, X.; Chen, J. Expression of Sonic Hedgehog Signaling Components in Hepatocellular Carcinoma and Cyclopamine-Induced Apoptosis Through Bcl-2 Downregulation In Vitro. Arch. Med. Res. 2010, 41, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Duman-Scheel, M.; Weng, L.; Xin, S.; Du, W. Hedgehog Regulates Cell Growth and Proliferation by Inducing Cyclin D and Cyclin, E. Nature 2002, 417, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Farah, M.H.; Eberhart, C.G. Hedgehog Signaling Promotes Medulloblastoma Survival via BclII. Am. J. Pathol. 2007, 170, 347–355. [Google Scholar] [CrossRef]
- Pola, R.; Ling, L.E.; Silver, M.; Corbley, M.J.; Kearney, M.; Blake Pepinsky, R.; Shapiro, R.; Taylor, F.R.; Baker, D.P.; Asahara, T.; et al. The Morphogen Sonic Hedgehog Is an Indirect Angiogenic Agent Upregulating Two Families of Angiogenic Growth Factors. Nat. Med. 2001, 7, 706–711. [Google Scholar] [CrossRef]
- Chen, M.; Qian, Y.; Dai, J.; Chu, R. The Sonic Hedgehog Signaling Pathway Induces Myopic Development by Activating Matrix Metalloproteinase (MMP)-2 in Guinea Pigs. PLoS ONE 2014, 9, e96952. [Google Scholar] [CrossRef]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of Hedgehog Signaling Inhibits Pancreatic Cancer Invasion and Metastases: A New Paradigm for Combination Therapy in Solid Cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef]
- Mazumdar, T.; Sandhu, R.; Qadan, M.; DeVecchio, J.; Magloire, V.; Agyeman, A.; Li, B.; Houghton, J.A. Hedgehog Signaling Regulates Telomerase Reverse Transcriptase in Human Cancer Cells. PLoS ONE 2013, 8, e75253. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Mack, S.C.; Mak, T.H.; Angers, S.; Taylor, M.D.; Hui, C.-C. Evasion of P53 and G2/M Checkpoints Are Characteristic of Hh-Driven Basal Cell Carcinoma. Oncogene 2014, 33, 2674–2680. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.M.; Ye, H.; Wetmore, C.; Karnitz, L.M. Sonic Hedgehog Signaling Impairs Ionizing Radiation–Induced Checkpoint Activation and Induces Genomic Instability. J. Cell Biol. 2008, 183, 385–391. [Google Scholar] [CrossRef]
- Scheffold, A.; Baig, A.H.; Chen, Z.; von Löhneysen, S.E.; Becker, F.; Morita, Y.; Avila, A.I.; Groth, M.; Lechel, A.; Schmid, F.; et al. Elevated Hedgehog Activity Contributes to Attenuated DNA Damage Responses in Aged Hematopoietic Cells. Leukemia 2020, 34, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Dreier, J.; Cheng, P.F.; Nägeli, M.; Lehmann, H.; Felderer, L.; Frew, I.J.; Matsushita, S.; Levesque, M.P.; Dummer, R. Hedgehog Pathway Inhibitors Promote Adaptive Immune Responses in Basal Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Merchant, J.L.; Ding, L. Hedgehog Signaling Links Chronic Inflammation to Gastric Cancer Precursor Lesions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog Pathway Activation in Cancer and Implications for Therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the Hedgehog Pathway in Patients with the Basal-Cell Nevus Syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Borrego, M.; Jimenez, B.; Antolín, S.; García-Saenz, J.A.; Corral, J.; Jerez, Y.; Trigo, J.; Urruticoechea, A.; Colom, H.; Gonzalo, N.; et al. A Phase Ib Study of Sonidegib (LDE225), an Oral Small Molecule Inhibitor of Smoothened or Hedgehog Pathway, in Combination with Docetaxel in Triple Negative Advanced Breast Cancer Patients: GEICAM/2012–12 (EDALINE) Study. Investig. New Drugs 2019, 37, 98–108. [Google Scholar] [CrossRef] [PubMed]
- NCT04007744; Sonidegib and Pembrolizumab in Treating Patients With Advanced Solid Tumors. National Library of Medicine: Bethesda, MD, USA; Mayo Clinic: Rochester, MN, USA, 2019.
- NCT03784014; Molecular Profiling of Advanced Soft-Tissue Sarcomas (MULTISARC). National Library of Medicine: Bethesda, MD, USA; Institut National de la Santé Et de la Recherche Médicale: Paris, France, 2018.
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glyn, M.; Zaphiropoulos, P.G.; Undén, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; Toftgård, R. The Role of the Human Homologue of Drosophila Patched in Sporadic Basal Cell Carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef]
- Yan, T.; Angelini, M.; Alman, B.A.; Andrulis, I.L.; Wunder, J.S. PATCHED-ONE or SMOOTHENED Gene Mutations Are Infrequent in Chondrosarcoma. Clin. Orthop. 2008, 466, 2184–2189. [Google Scholar] [CrossRef]
- Wang, X.-D.; Inzunza, H.; Chang, H.; Qi, Z.; Hu, B.; Malone, D.; Cogswell, J. Mutations in the Hedgehog Pathway Genes SMO and PTCH1 in Human Gastric Tumors. PLoS ONE 2013, 8, e54415. [Google Scholar] [CrossRef]
- Lam, C.-W.; Xie, J.; To, K.-F.; Ng, H.-K.; Lee, K.-C.; Yuen, N.W.-F.; Lim, P.-L.; Chan, L.Y.-S.; Tong, S.-F.; McCormick, F. A Frequent Activated Smoothened Mutation in Sporadic Basal Cell Carcinomas. Oncogene 1999, 18, 833–836. [Google Scholar] [CrossRef]
- Ighilahriz, M.; Nikolaev, S.; Yurchenko, A.; Battistella, M.; Mourah, S.; Jouenne, F.; Bourrat, E.; Basset-Seguin, N. A Novel Case of Gorlin Syndrome Mosaicism Involving an SMO Gene Mutation: Clinical, Histological and Molecular Analysis of Basaloid Tumours. Acta Derm. Venereol. 2021, 101, adv00434. [Google Scholar] [CrossRef]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Ditzler, S.; Knoblaugh, S.E.; Hatton, B.A.; Schelter, J.M.; Cleary, M.A.; Mecham, B.; Rorke-Adams, L.B.; Olson, J.M. A Distinct Smoothened Mutation Causes Severe Cerebellar Developmental Defects and Medulloblastoma in a Novel Transgenic Mouse Model. Mol. Cell. Biol. 2012, 32, 4104–4115. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The Whole-Genome Landscape of Medulloblastoma Subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Boetto, J.; Bielle, F.; Sanson, M.; Peyre, M.; Kalamarides, M. SMO Mutation Status Defines a Distinct and Frequent Molecular Subgroup in Olfactory Groove Meningiomas. Neuro-Oncol. 2017, 19, 345–351. [Google Scholar] [CrossRef]
- Thomas, X.; Heiblig, M. An Evaluation of Glasdegib for the Treatment of Acute Myelogenous Leukemia. Expert Opin. Pharmacother. 2020, 21, 523–530. [Google Scholar] [CrossRef]
- Chang, A.L.S.; Arron, S.T.; Migden, M.R.; Solomon, J.A.; Yoo, S.; Day, B.-M.; McKenna, E.F.; Sekulic, A. Safety and Efficacy of Vismodegib in Patients with Basal Cell Carcinoma Nevus Syndrome: Pooled Analysis of Two Trials. Orphanet J. Rare Dis. 2016, 11, 120. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Chang, A.L.S.; Oro, A.E. Initial Assessment of Tumor Regrowth After Vismodegib in Advanced Basal Cell Carcinoma. Arch. Dermatol. 2012, 148, 1324. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef]
- Pricl, S.; Cortelazzi, B.; Dal Col, V.; Marson, D.; Laurini, E.; Fermeglia, M.; Licitra, L.; Pilotti, S.; Bossi, P.; Perrone, F. Smoothened (SMO) Receptor Mutations Dictate Resistance to Vismodegib in Basal Cell Carcinoma. Mol. Oncol. 2015, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L.S. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.W.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef]
- NCT04341181; ProTarget-A Danish Nationwide Clinical Trial on Targeted Cancer Treatment Based on Genomic Profiling (ProTarget). National Library of Medicine: Bethesda, MD, USA, 2020.
- NCT05159245; The Finnish National Study to Facilitate Patient Access to Targeted Anti-Cancer Drugs (FINPROVE). National Library of Medicine: Bethesda, MD, USA; Helsinki University Central Hospital: Helsinki, Finland, 2021.
- Zhang, H.; Sun, Z.; Liu, Z.; Song, C. Overcoming the Emerging Drug Resistance of Smoothened: An Overview of Small-Molecule SMO Antagonists with Antiresistance Activity. Future Med. Chem. 2018, 10, 2855–2875. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-W.; Yarravarapu, N.; Shi, H.; Kulak, O.; Kim, J.; Chen, C.; Lum, L. A Synthetic Combinatorial Approach to Disabling Deviant Hedgehog Signaling. Sci. Rep. 2018, 8, 1133. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Gouti, M.; Sagner, A.; Sircar, R.; Luchetti, G.; Ingham, P.W.; Briscoe, J.; Rohatgi, R. G Protein–Coupled Receptors Control the Sensitivity of Cells to the Morphogen Sonic Hedgehog. Sci. Signal. 2018, 11, eaao5749. [Google Scholar] [CrossRef]
- Syed Haneef, S.A.; Ranganathan, S. Structural Bioinformatics Analysis of Variants on GPCR Function. Curr. Opin. Struct. Biol. 2019, 55, 161–177. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Indications | Stage |
|---|---|---|
| IP-926 (Saridegib) | Metastatic pancreatic cancer (NCT01130142) Myelofibrosis (NCT01371617) Chondrosarcoma (NCT01310816) BCC (NCT01609179, NCT02828111) BCN (NCT02762084, NCT03703310) | Phase II, III |
| BMS-833923/XL139 | BCC and BCN (NCT00670189) Chronic Myeloid Leukaemia (NCT01218477) Small Cell Lung Cancer (NCT00927875) Gastric and Oesophageal Adenocarcinomas (NCT00909402) Multiple myeloma (NCT00884546) | Phase I, II |
| LY2940680 (Taladegib) | Advanced solid tumours (NCT02784795) Small Cell Lung Cancer (NCT01722292) Gastroesophageal Junction Adenocarcinoma (NCT02530437) | Phase I, II |
| Vitamin D3 | BCC (NCT01358045) | Phase II |
| Itraconazole | BCC (NCT02120677) Non-Small Cell Lung Cancer (NCT02357836) Prostate Cancer (NCT01787331) Oesophageal Squamous Cell Carcinoma (NCT04018872) | Phase II |
| LEQ-506 | Advanced solid tumours (NCT01106508) | Phase I |
| Tumour | Driving Mutations |
|---|---|
| Basal cell carcinoma | R562Q, W535L |
| Basal cell nevus syndrome | L412F |
| Hepatocellular carcinoma | K575M |
| Medulloblastoma | W535L, S533N, S278I |
| Meningioma | L412F, W535L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicheperovich, A.; Townsend-Nicholson, A. Towards Precision Oncology: The Role of Smoothened and Its Variants in Cancer. J. Pers. Med. 2022, 12, 1648. https://doi.org/10.3390/jpm12101648
Nicheperovich A, Townsend-Nicholson A. Towards Precision Oncology: The Role of Smoothened and Its Variants in Cancer. Journal of Personalized Medicine. 2022; 12(10):1648. https://doi.org/10.3390/jpm12101648
Chicago/Turabian StyleNicheperovich, Alina, and Andrea Townsend-Nicholson. 2022. "Towards Precision Oncology: The Role of Smoothened and Its Variants in Cancer" Journal of Personalized Medicine 12, no. 10: 1648. https://doi.org/10.3390/jpm12101648
APA StyleNicheperovich, A., & Townsend-Nicholson, A. (2022). Towards Precision Oncology: The Role of Smoothened and Its Variants in Cancer. Journal of Personalized Medicine, 12(10), 1648. https://doi.org/10.3390/jpm12101648