
The Effect of Radiotherapy on Diffuse Low-Grade Gliomas Evolution: Confronting Theory with Clinical Data

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Patients
2.2. Standard Protocol Approvals, Registration, and Patient Consent
2.3. The Model
2.3.1. Free Tumor Evolution
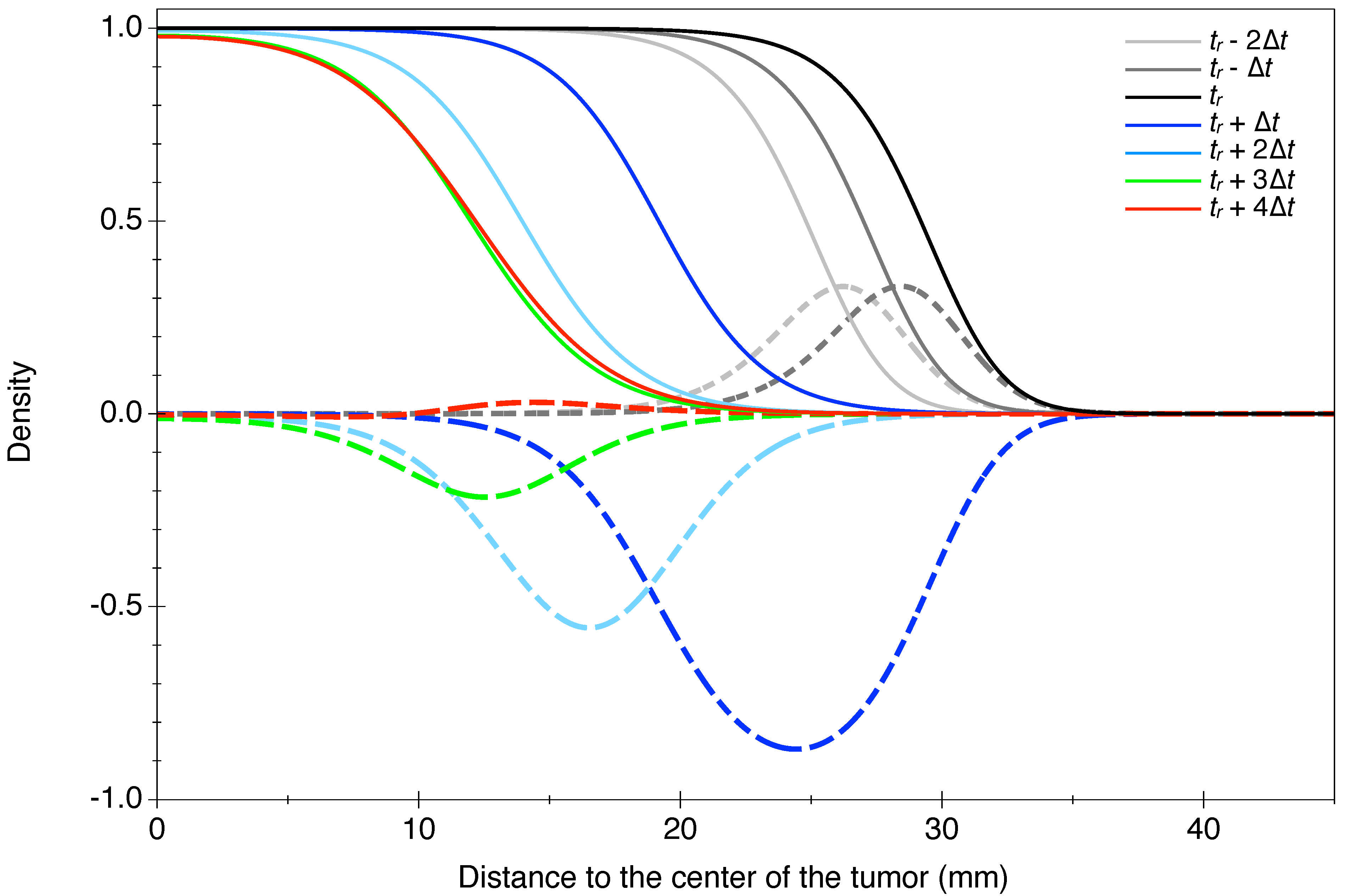
2.3.2. Modeling RT
2.4. Fitting Procedure
3. Results
3.1. Characterization of Our Model
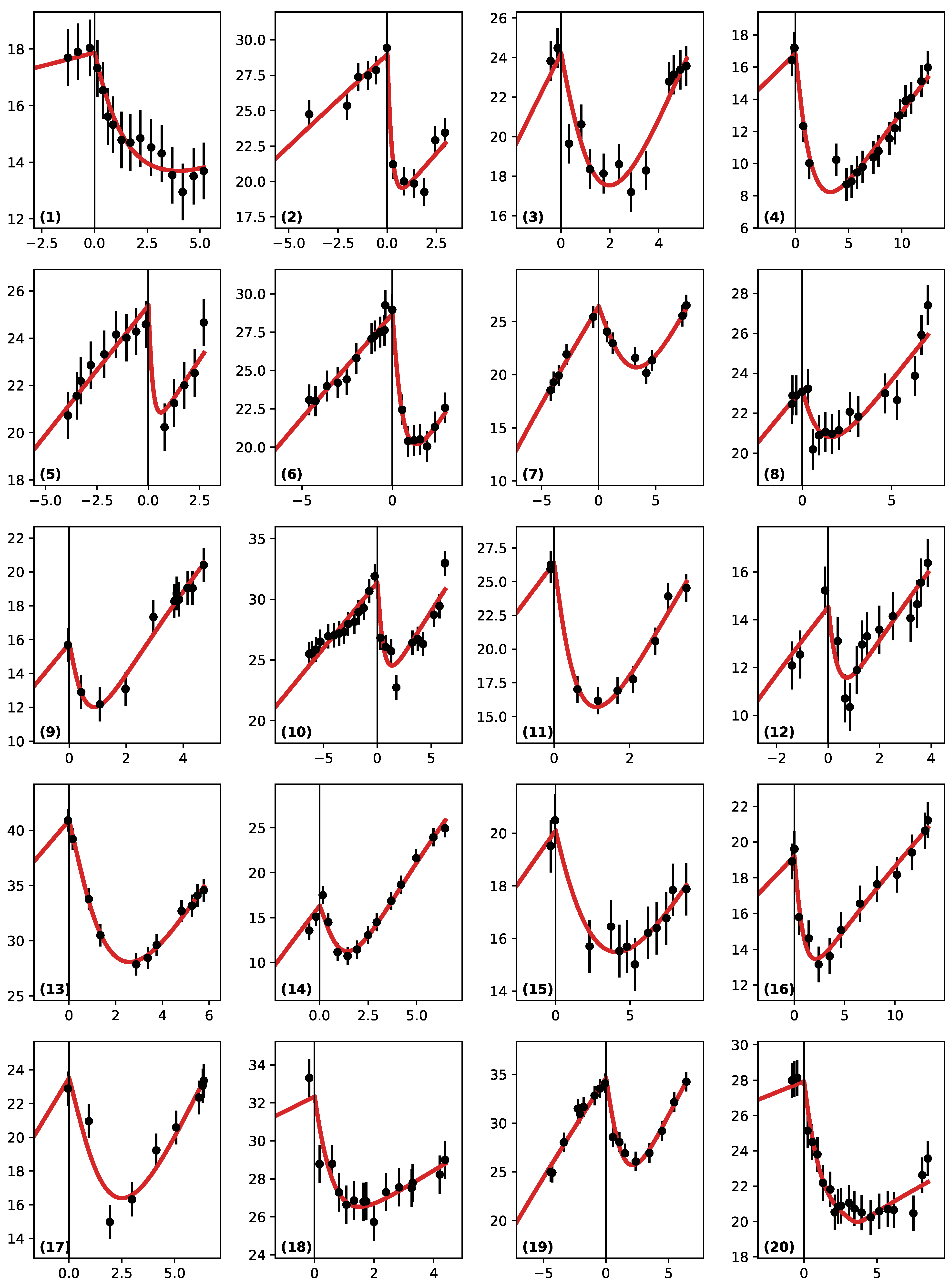
3.2. Best Fits
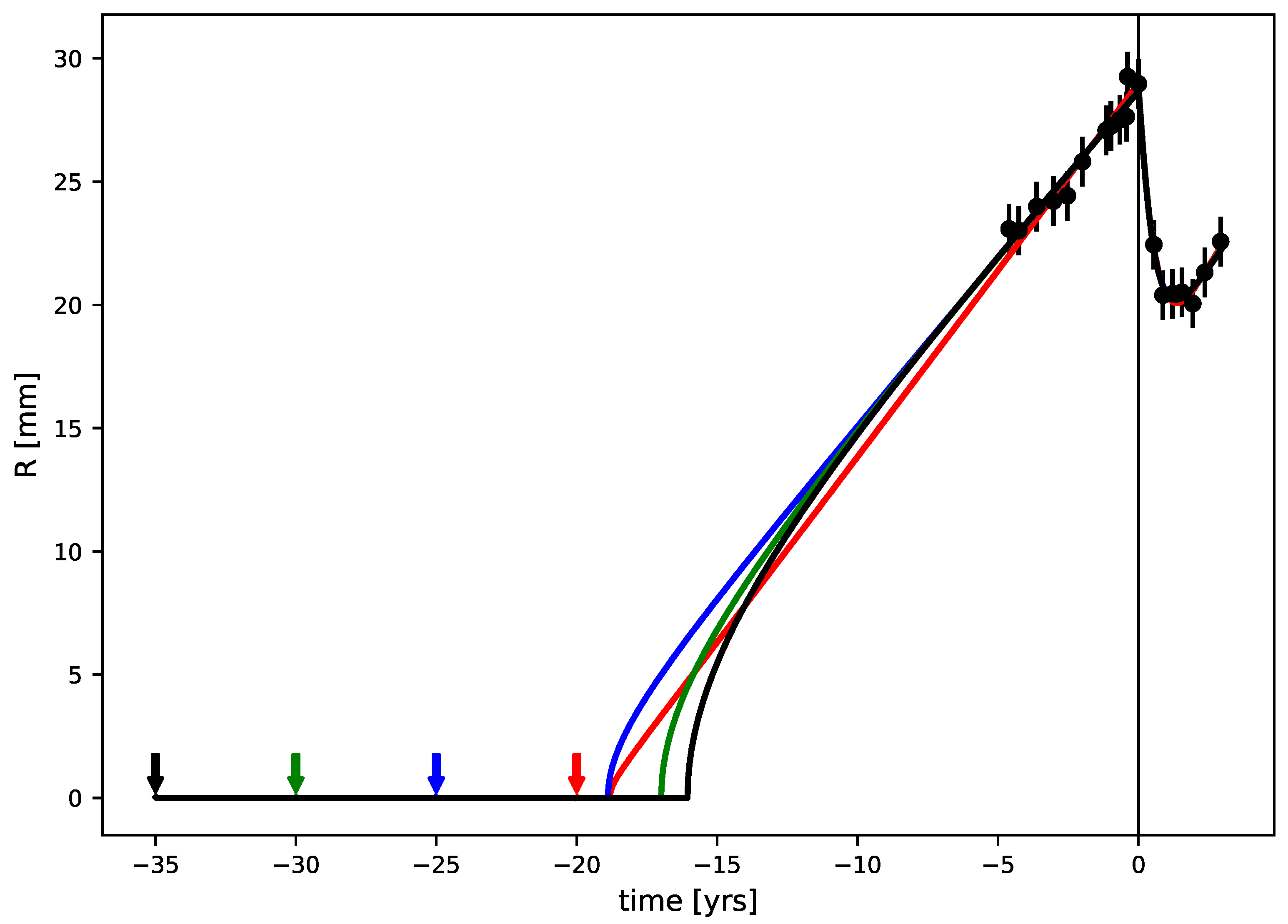
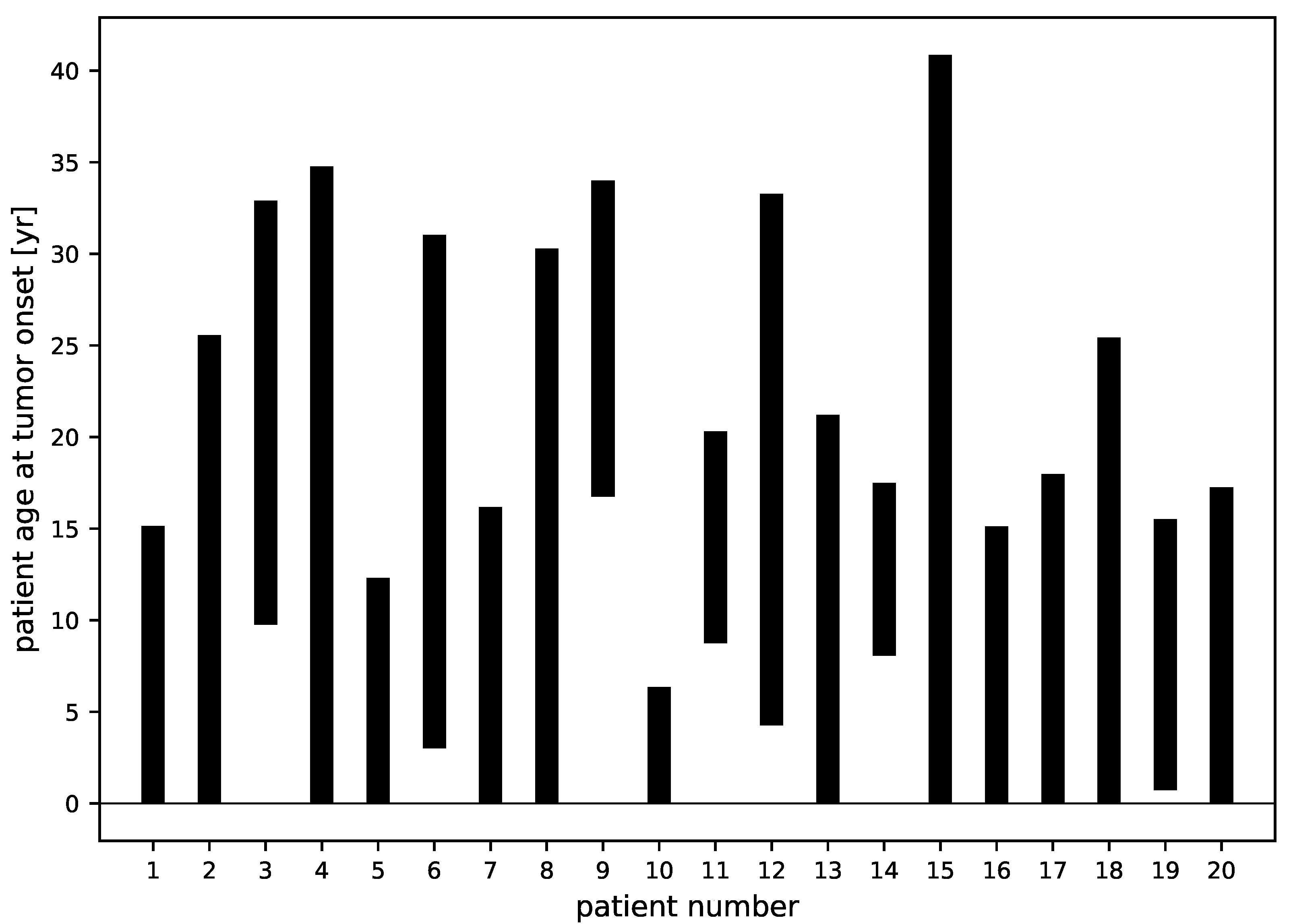
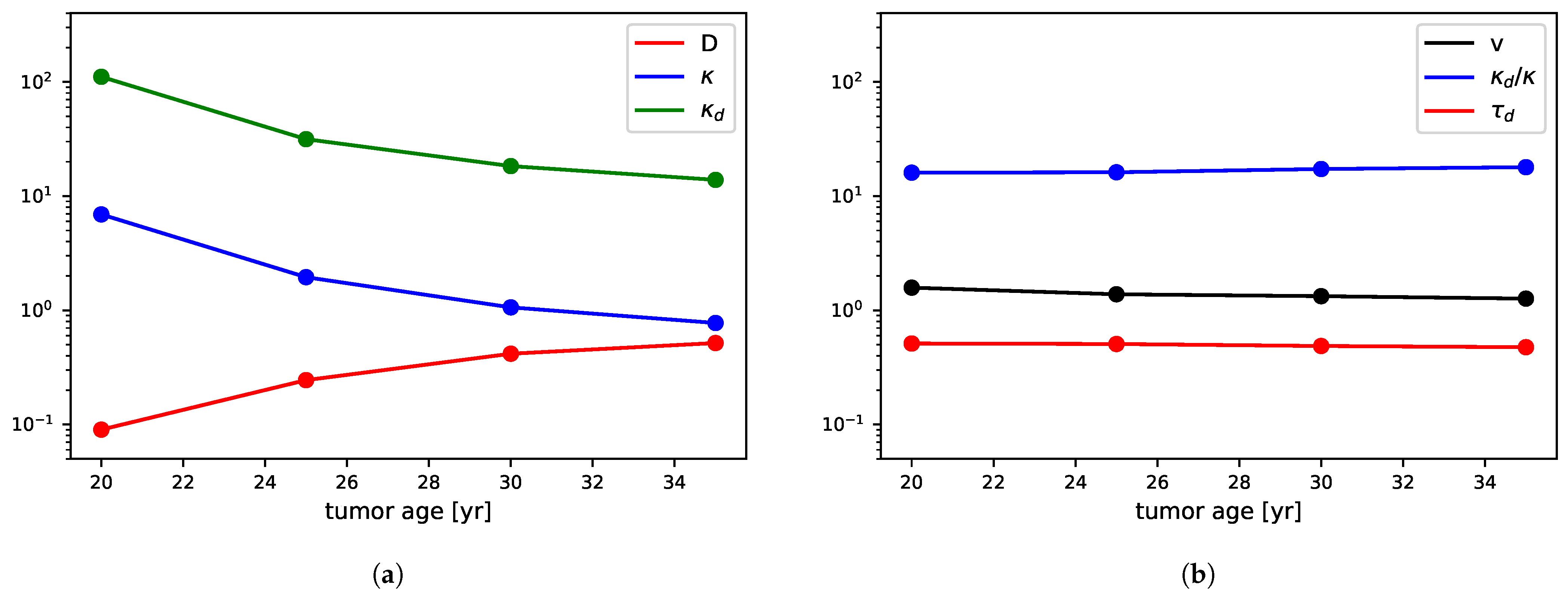
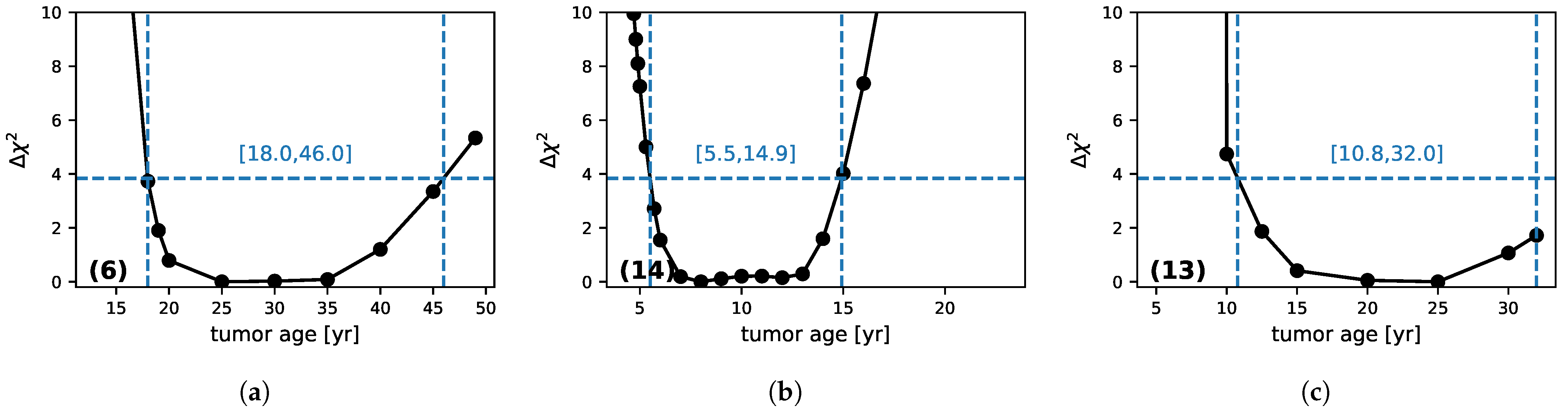
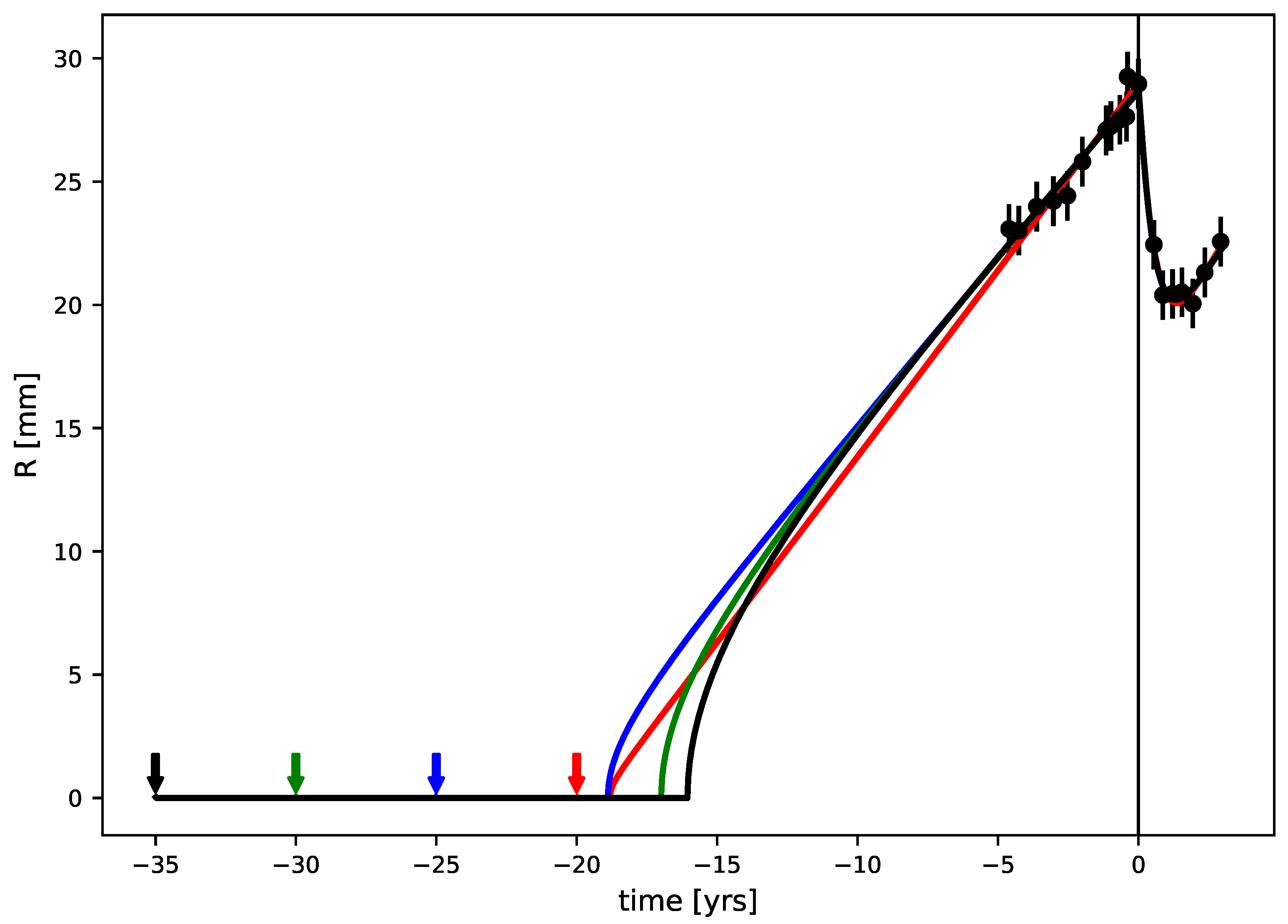
3.3. Tumor Age
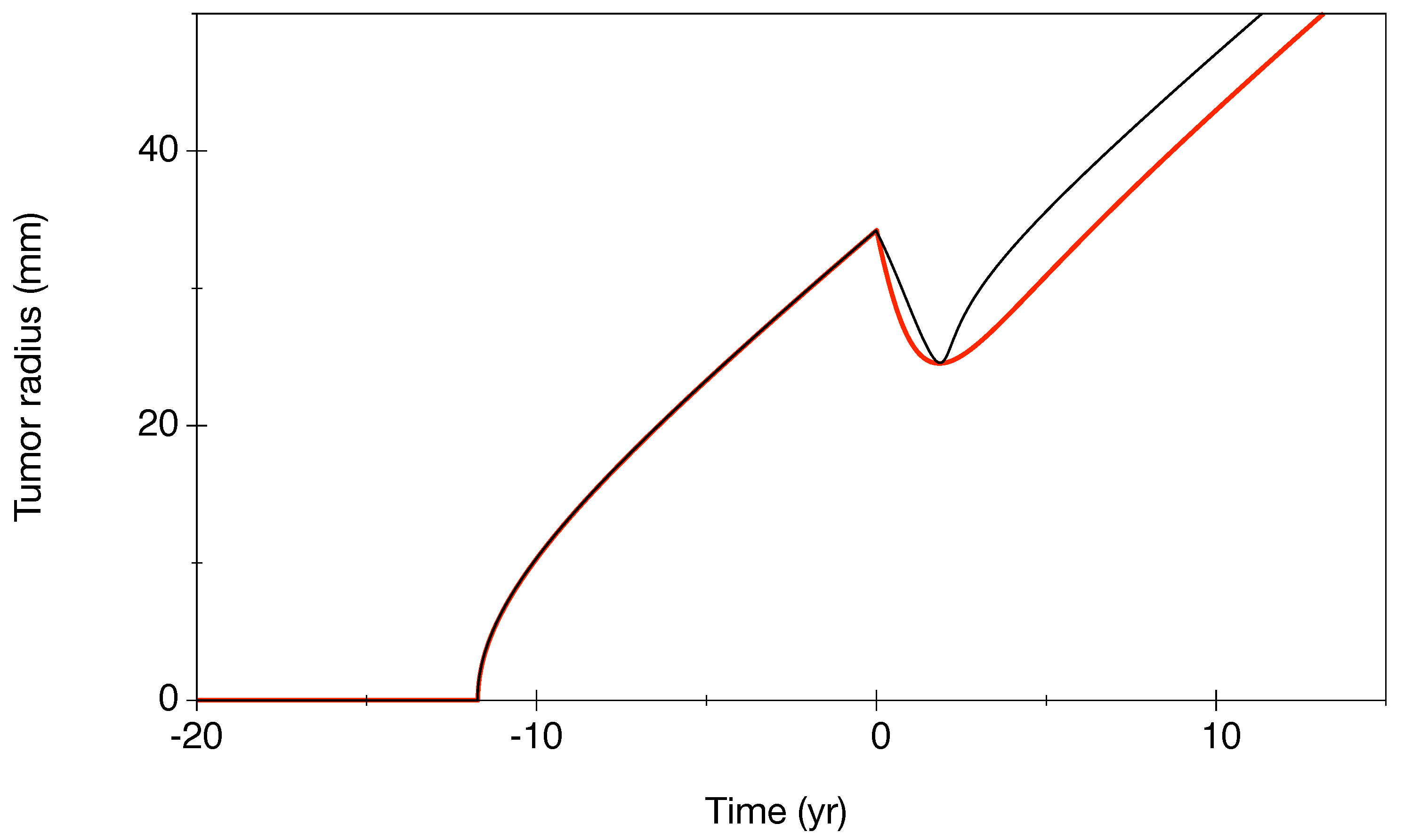
- (a): Patient (6): This is a case where there are many points before RT and few during the regrowth phase.
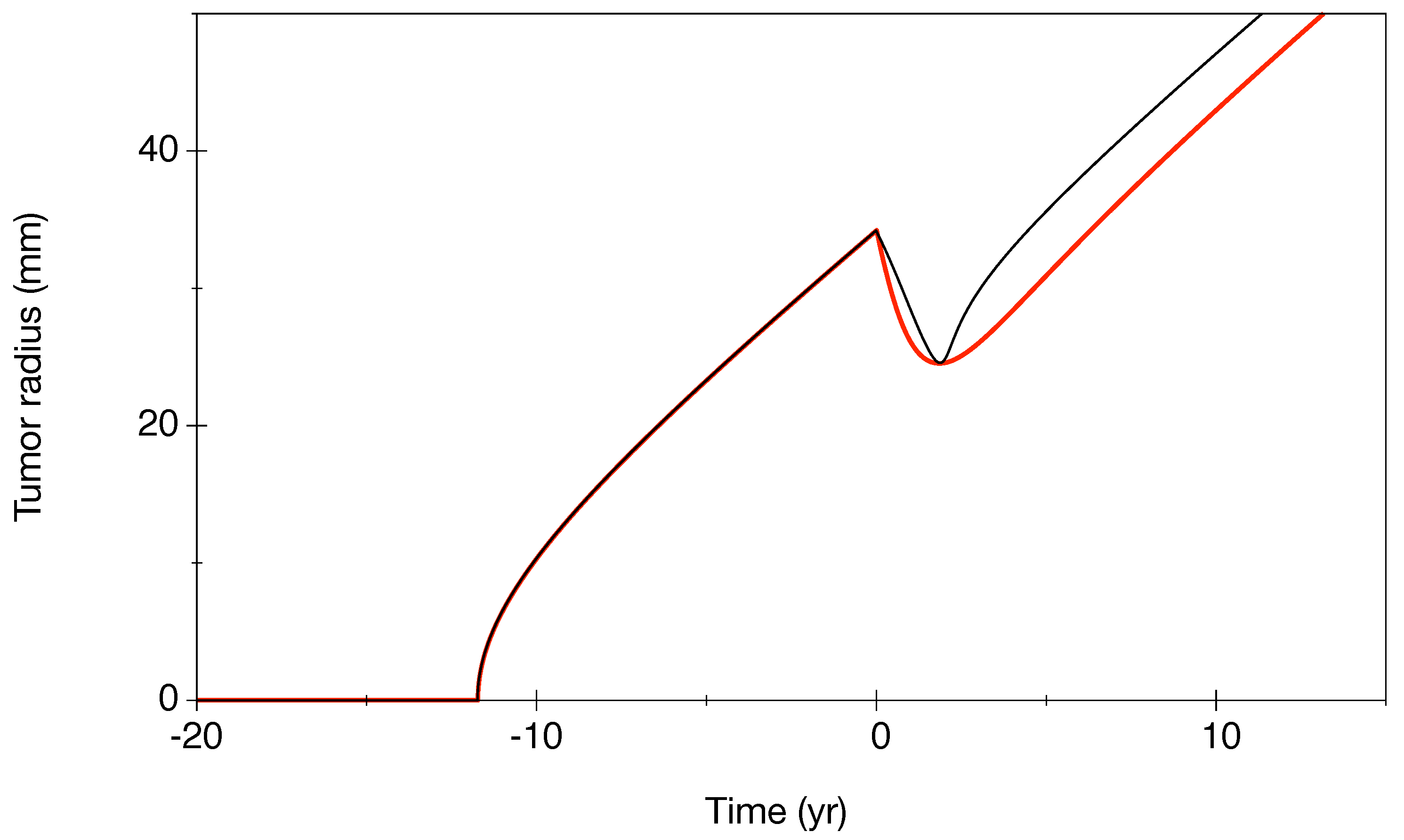
- (b): Patient (14) is the inverse, with few points before RT, but the regrowth is strongly sampled.
- (c): Patient (13): No points before RT and a few during regrowth.
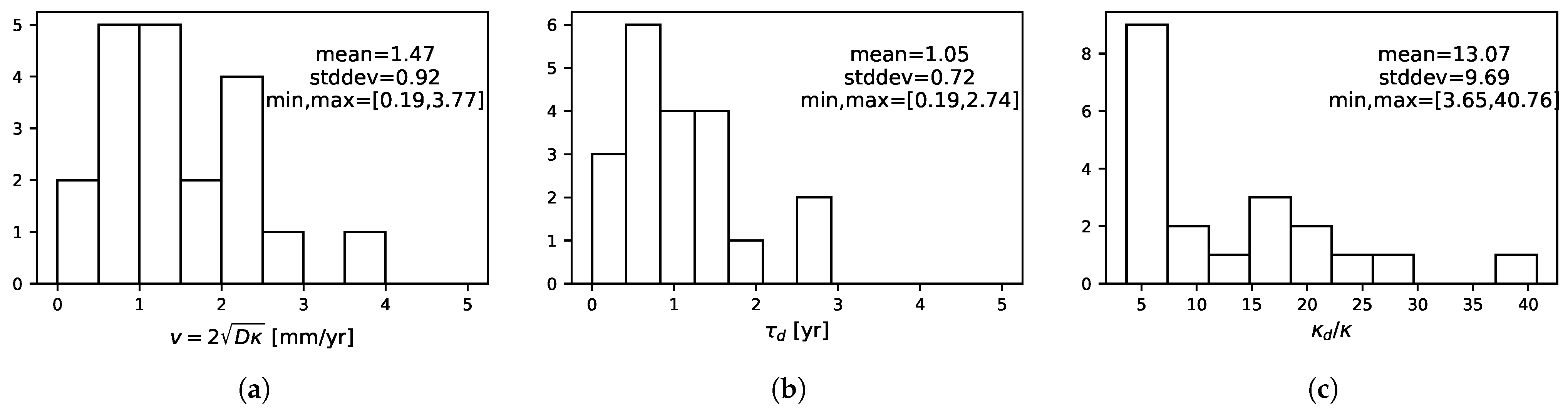
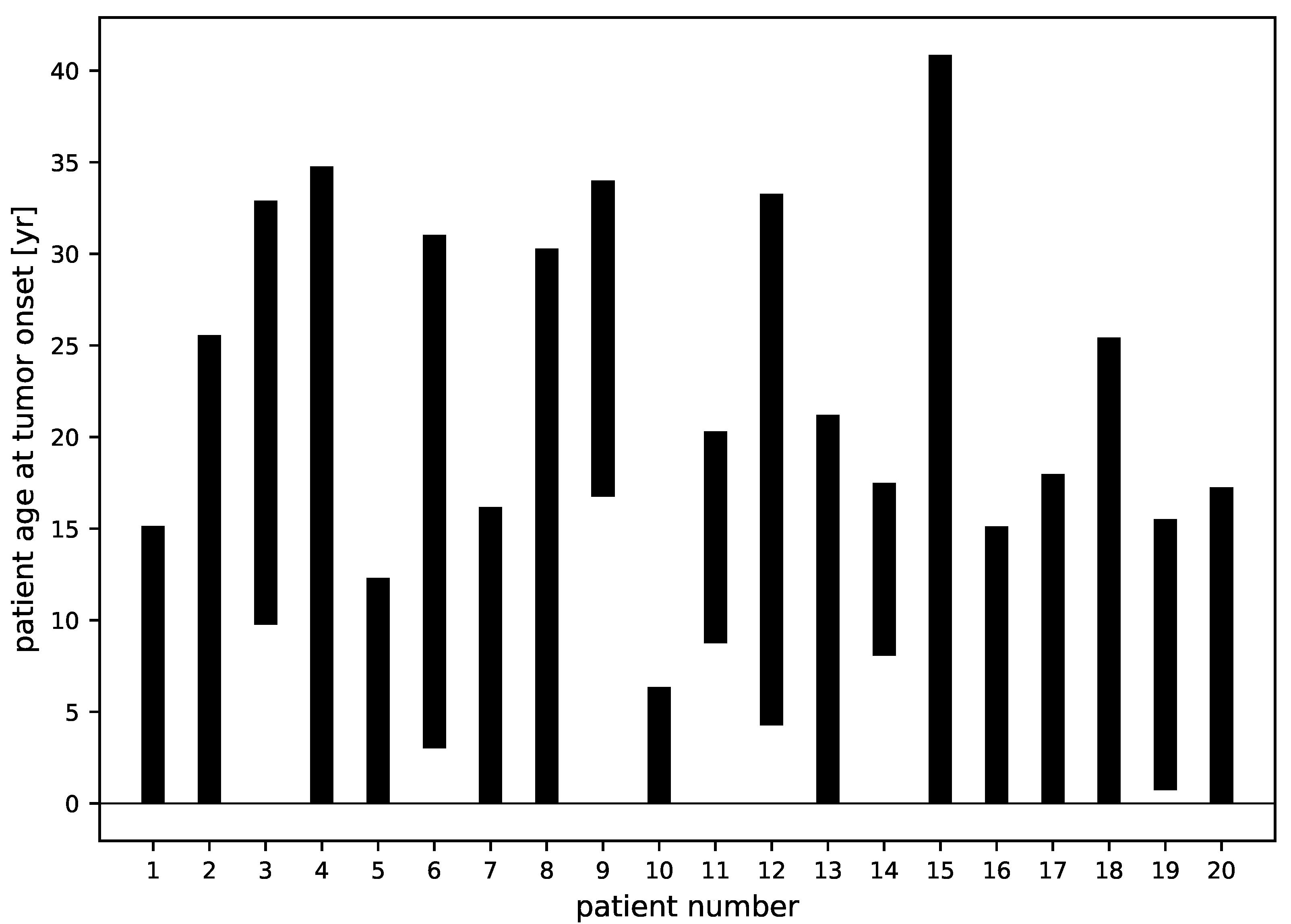
3.4. Tumor Characteristics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Appendix A
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.; Ohgaki, H.; Wiestler, O.; Kleihues, P.; Ellison, D. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallud, J.; Llitjos, J.F.; Dhermain, F.; Varlet, P.; Dezamis, E.; Devaux, B.; Souillard-Scémama, R.; Sanai, N.; Koziak, M.; Page, P.; et al. Dynamic imaging response following radiation therapy predicts long-term outcomes for diffuse low-grade gliomas. Neuro Oncol. 2012, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pallud, J.; Mandonnet, E. Quantitative Approach of the Natural Course of Diffuse Low-Grade Gliomas. In Tumors of the Central Nervous System; Springer: Dordrecht, The Netherlands, 2011; Volume 2, pp. 163–172. [Google Scholar]
- Price, S.J. Advances in imaging low-grade gliomas. Adv. Tech. Stand. Neurosurg 2010, 35, 1–34. [Google Scholar]
- Kelly, P.J.; Daumas-Duport, C.; Kispert, D.B.; Kall, B.A.; Scheithauer, W.; Illig, J. Imaging-based stereotaxic serial biopsies in untreated intracranial glial neaplasms. J. Neurosurg. 1987, 66, 865–874. [Google Scholar] [CrossRef]
- Pallud, J.; Varlet, P.; Devaux, B.; Geha, S.; Badoual, M.; Deroulers, C.; Page, P.; Dezamis, E.; Daumas-Duport, C.; Roux, F.X. Diffuse low-grade oligodendrogliomas extend beyond MRI-defined abnormalities. Neurology 2010, 74, 1724–1731. [Google Scholar] [CrossRef]
- Pallud, J.; Fontaine, D.; Duffau, H.; Mandonnet, E.; Sanai, N.; Taillandier, L.; Peruzzi, P.; Guillevin, R.; Bauchet, L.; Bernier, V.; et al. Natural history of incidental WHO grade II gliomas. Ann. Neurol. 2010, 68, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Ricard, D.; Kaloshi, G.; Amiel-Benouaich, A.; Lejeune, J.; Marie, Y.; Mandonnet, E.; Kujas, M.; Mokhtari, K.; Taillibert, S.; Laigle-Donadey, F.; et al. Dynamic history of low-grade gliomas before and after temozolomide treatment. Ann. Neurol. 2007, 61, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Peyre, M.; Cartalat-Carel, S.; Meyronet, D.; Ricard, D.; Jouvet, A.; Pallud, J.; Mokhtari, K.; Guyotat, J.; Jouanneau, E.; Sunyach, M.; et al. Prolonged response without prolonged chemotherapy: A lesson from PCV chemotherapy in low-grade gliomas. Neuro. Oncol. 2010, 12, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Mandonnet, E.; Pallud, J.; Fontaine, D.; Taillandier, L.; Bauchet, L.; Peruzzi, P.; Guyotat, J.; Bernier, V.; Baron, M.H.; Duffau, H.; et al. Inter- and intrapatients comparison of WHO grade II glioma kinetics before and after surgical resection. Neurosurg. Rev. 2009, 33, 91–96. [Google Scholar] [CrossRef]
- Prabhu, V.C.; Khaldi, A.; Barton, K.P.; Melian, E.; Schneck, M.J.; Primeau, M.J.; Lee, J.M. Management of diffuse low-grade cerebral gliomas. Neurol. Clin. 2010, 28, 1037–1059. [Google Scholar] [CrossRef]
- Bobek-Billewicz, B.; Stasik-Pres, G.; Hebda, A.; Majchrzak, K.; Kaspera, W.; Jurkowski, M. Anaplastic transformation of low-grade gliomas (WHO II) on magnetic resonance imaging. Folia Neuropathol. 2014, 52, 128–140. [Google Scholar] [CrossRef] [Green Version]
- van den Bent, M.; Afra, D.; de Witte, O.; Ben Hassel, M.; Schraub, S.; Hoang-Xuan, K.; Malmström, P.; Collette, L.; Piérart, M.; Mirimanoff, R.; et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: The EORTC 22845 randomised trial. Lancet 2005, 366, 985–990. [Google Scholar] [CrossRef]
- Dufour, A.; Gontran, E.; Deroulers, C.; Varlet, P.; Pallud, J.; Grammaticos, B.; Badoual, M. Modeling the dynamics of oligodendrocyte precursor cells and the genesis of gliomas. Cancer 2018, 14, e1005977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerin, C.; Pallud, J.; Grammaticos, B.; Mandonnet, E.; Deroulers, C.; Varlet, P.; Capelle, L.; Taillandier, L.; Bauchet, L.; Duffau, H.; et al. Improving the time-machine: Estimating date of birth of grade II gliomas. Cell. Prolif. 2012, 45, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Ribba, B.; Kaloshi, G.; Peyre, M.; Ricard, D.; Calvez, V.; Tod, M.; Cajavec-Bernard, B.; Idbaih, A.; Psimaras, D.; Dainese, L.; et al. A tumor growth inhibition model for low-grade glioma treated with chemotherapy or radiotherapy. Clin. Cancer Res. 2012, 18, 5071–5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badoual, M.; Gerin, C.; Deroulers, C.; Grammaticos, B.; Llitjos, J.; Oppenheim, C.; Varlet, P.; Pallud, J. Oedema-based model for diffuse low-grade gliomas: Application to clinical cases under radiotherapy. Cell. Prolif. 2014, 47, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Perez-García, V.; Bogdanska, M.; Martínez-Gonzalez, A.; Belmonte-Beitia, J.; Schucht, P.; Perez-Romasanta, L. Delay effects in the response of low-grade gliomas to radiotherapy: A mathematical model and its therapeutical implications. Math. Med. Biol. 2015, 32, 307–329. [Google Scholar] [CrossRef] [Green Version]
- Galochkina, T.; Bratus, A.; Perez-Garcia, V. Optimal radiation fractionation for low-grade gliomas: Insights from a mathematical model. Math. Biosci. 2015, 267, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Budia, I.; Alvarez-Arenas, A.; Woolley, T.; Calvo, G.; Belmonte-Beitia, J. Radiation protraction schedules for low-grade gliomas: A comparison between different mathematical models. J. R. Soc. Interface 2019, 16, 20190665. [Google Scholar] [CrossRef] [Green Version]
- Bogdanska, M.; Bodnar, M.; Piotrowska, M.; Murek, M.; Schucht, P.; Beck, J.; Martinez-Gonzalez, A.; Perez-Garcia, V. A mathematical model describes the malignant transformation of low grade gliomas: Prognostic implications. PLoS ONE 2017, 12, e0179999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockne, R.; Rockhill, J.K.; Mrugala, M.; Spence, A.M.; Kalet, I.; Hendrickson, K.; Lai, A.; Cloughesy, T.; Alvord, E.C.J.; Swanson, K.R. Predicting the efficacy of radiotherapy in individual glioblastoma patients in vivo: A mathematical modelling approach. Phys. Med. Biol 2010, 55, 3271–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henares-Molina, A.; Benzekry, S.; Lara, P.; Garcia-Rojo, M.; Perez-Garcia, V.; Martinez-Gonzalez, A. Non-standard radiotherapy fractionations delay the time to malignant transformation of low-grade gliomas. PLoS ONE 2017, 12, e0178552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruywagen, G.C.; Woodward, D.E.; Tracqui, P.; Bartoo, G.T.; Murray, J.D.; Alvord, E.C. The modelling of diffusive tumours. J. Biol. Syst. 1995, 3, 937–945. [Google Scholar] [CrossRef]
- Swanson, K.R.; Alvord, E.C.; Murray, J.D. A quantitative model for differential motility of gliomas in grey and white matter. Cell Prolif. 2000, 33, 317–329. [Google Scholar] [CrossRef]
- Harpold, H.L.; Alvord, E.C., Jr.; Swanson, K.R. The evolution of mathematical modeling of glioma proliferation and invasion. J. Neuropathol. Exp. Neurol. 2007, 1, 1–9. [Google Scholar] [CrossRef]
- Badoual, M.; Deroulers, C.; Aubert, M.; Grammaticos, B. Modelling intercellular communication and its effect on tumour invasion. Phys. Biol. 2010, 7, 046013. [Google Scholar] [CrossRef]
- Corwin, D.; Holdsworth, C.; Rockne, R.; Trister, A.; Mrugala, M.; Rockhill, J.; Stewart, R.; Phillips, M.; Swanson, K. Toward patient-specific, biologically optimized radiation therapy plans for the treatment of glioblastoma. PLoS ONE 2013, 8, e79115. [Google Scholar] [CrossRef]
- Unkelbach, J.; Menze, B.; Konukoglu, E.; Dittmann, F.; Le, M.; Ayache, N.; Shih, H. Radiotherapy planning for glioblastoma based on a tumor growth model: improving target volume delineation. Phys. Med. Biol. 2014, 59, 747–770. [Google Scholar] [CrossRef] [Green Version]
- Amelot, A.; Deroulers, C.; Badoual, M.; Polivka, M.; Adle-Biassette, H.; Houdart, E.; Carpentier, A.; Froelich, S.; Mandonnet, E. Surgical Decision Making From Image-Based Biophysical Modeling of Glioblastoma: Not Ready for Primetime. Neurosurgery 2017, 80, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Mandonnet, E.; Delattre, J.Y.; Tanguy, M.L.; Swanson, K.R.; Carpentier, A.F.; Duffau, H.; Cornu, P.; Van Effenterre, R.; Alvord, E.C., Jr.; Capelle, L. Continuous growth of mean tumor diameter in a subset of grade II gliomas. Ann. Neurol. 2003, 53, 524–528. [Google Scholar] [CrossRef]
- Pallud, J.; Mandonnet, E.; Duffau, H.; Kujas, M.; Guillevin, R.; Galanaud, D.; Taillandier, L.; Capelle, L. Prognostic value of initial magnetic resonance imaging growth rates for World Health Organization grade II gliomas. Ann. Neurol. 2006, 60, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Hansen, N.; Ostermeier, A. Completely Derandomized Self-Adaptation in Evolution Strategies. Evol. Comput. 2001, 9, 159–195. [Google Scholar] [CrossRef]
- Murray, J.D. Mathematical Biology. II: Spatial Models and Biomedical Applications, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Pallud, J.; Capelle, L.; Taillandier, L.; Badoual, M.; Duffau, H.; Mandonnet, E. The silent phase of diffuse low-grade gliomas. Is it when we missed the action? Acta Neurochir. 2013, 155, 2237–2242. [Google Scholar] [CrossRef]
- James, F. Statistical Methods in Experimental Physics; World Scientific: Singapour, 2007. [Google Scholar]
- James, F. Interpretation of the shape of the likelihood function around its minimum. Comput. Phys. Commun. 1980, 20, 29–35. [Google Scholar] [CrossRef]
- Mandonnet, E.; Pallud, J.; Clatz, O.; Taillandier, L.; Konukoglu, E.; Duffau, H.; Capelle, L. Computational modeling of the WHO grade II glioma dynamics: principles and applications to management paradigm. Neurosurg. Rev 2008, 31, 263–269. [Google Scholar] [CrossRef] [PubMed]
- El-Hateer, H.; Souhami, L.; Roberge, D.; Maestro, R.D.; Leblanc, R.; Eldebawy, E.; Muanza, T.; Melançon, D.; Kavan, P.; Guiot, M.C. Low-grade oligodendroglioma: An indolent but incurable disease? J. Neurosurg. 2009, 111, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Gerin, C.; Pallud, J.; Deroulers, C.; Varlet, P.; Oppenheim, C.; Roux, F.X.; Chrétien, F.; Grammaticos, B.; Badoual, M. Quantitative characterization of the imaging limits of diffuse low-grade oligodendrogliomas. Neuro Oncol. 2013, 15, 1379–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.J.; Ngok, Y. Response of cells to ionizing radiation. Avd. Biomed. Sci. Eng. 2009, 59, 204–262. [Google Scholar]
- Wang, J.; Wang, H.; Qian, H. Biological effects of radiation on cancer cells. Mil. Med. Res. 2018, 5, 20. [Google Scholar] [CrossRef]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell. Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- McMahon, S. The linear quadratic model: Usage, interpretation and challenges. Phys. Med. Biol. 2018, 64, 01TR01. [Google Scholar] [CrossRef] [PubMed]
- Amberger-Murphy, V. Hypoxia helps glioma to fight therapy. Curr. Cancer Drug Targets 2009, 9, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruningk, S.; Rivens, I.; Box, C.; Oelfke, U.; Ter Haar, G. 3D tumour spheroids for the prediction of the effects of radiation and hyperthermia treatments. Sci. Rep. 2020, 10, 1653. [Google Scholar] [CrossRef]
- Bruningk, S.; Ziegenhein, P.; Rivens, I.; Oelfke, U.; Ter Haar, G. A cellular automaton model for spheroid response to radiation and hyperthermia treatments. Sci. Rep. 2019, 9, 17674. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adenis, L.; Plaszczynski, S.; Grammaticos, B.; Pallud, J.; Badoual, M. The Effect of Radiotherapy on Diffuse Low-Grade Gliomas Evolution: Confronting Theory with Clinical Data. J. Pers. Med. 2021, 11, 818. https://doi.org/10.3390/jpm11080818
Adenis L, Plaszczynski S, Grammaticos B, Pallud J, Badoual M. The Effect of Radiotherapy on Diffuse Low-Grade Gliomas Evolution: Confronting Theory with Clinical Data. Journal of Personalized Medicine. 2021; 11(8):818. https://doi.org/10.3390/jpm11080818
Chicago/Turabian StyleAdenis, Léo, Stéphane Plaszczynski, Basile Grammaticos, Johan Pallud, and Mathilde Badoual. 2021. "The Effect of Radiotherapy on Diffuse Low-Grade Gliomas Evolution: Confronting Theory with Clinical Data" Journal of Personalized Medicine 11, no. 8: 818. https://doi.org/10.3390/jpm11080818
APA StyleAdenis, L., Plaszczynski, S., Grammaticos, B., Pallud, J., & Badoual, M. (2021). The Effect of Radiotherapy on Diffuse Low-Grade Gliomas Evolution: Confronting Theory with Clinical Data. Journal of Personalized Medicine, 11(8), 818. https://doi.org/10.3390/jpm11080818