
DPYD Exome, mRNA Expression and Uracil Levels in Early Severe Toxicity to Fluoropyrimidines: An Extreme Phenotype Approach

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Ethics
2.3. DPYD Exon Sequencing
2.4. DPD Activity Measure
2.5. DPYD mRNA Expression
2.5.1. RNA Isolation cDNA Synthesis
2.5.2. Real-Time PCR and Quantification Method
2.6. Characterization of 2197insA
2.6.1. mRNA Sequencing
2.6.2. 3D Modeling
2.7. Analysis of Exon 4 Skipping
3. Results
3.1. Clinical and Demographic Characteristics of Patients
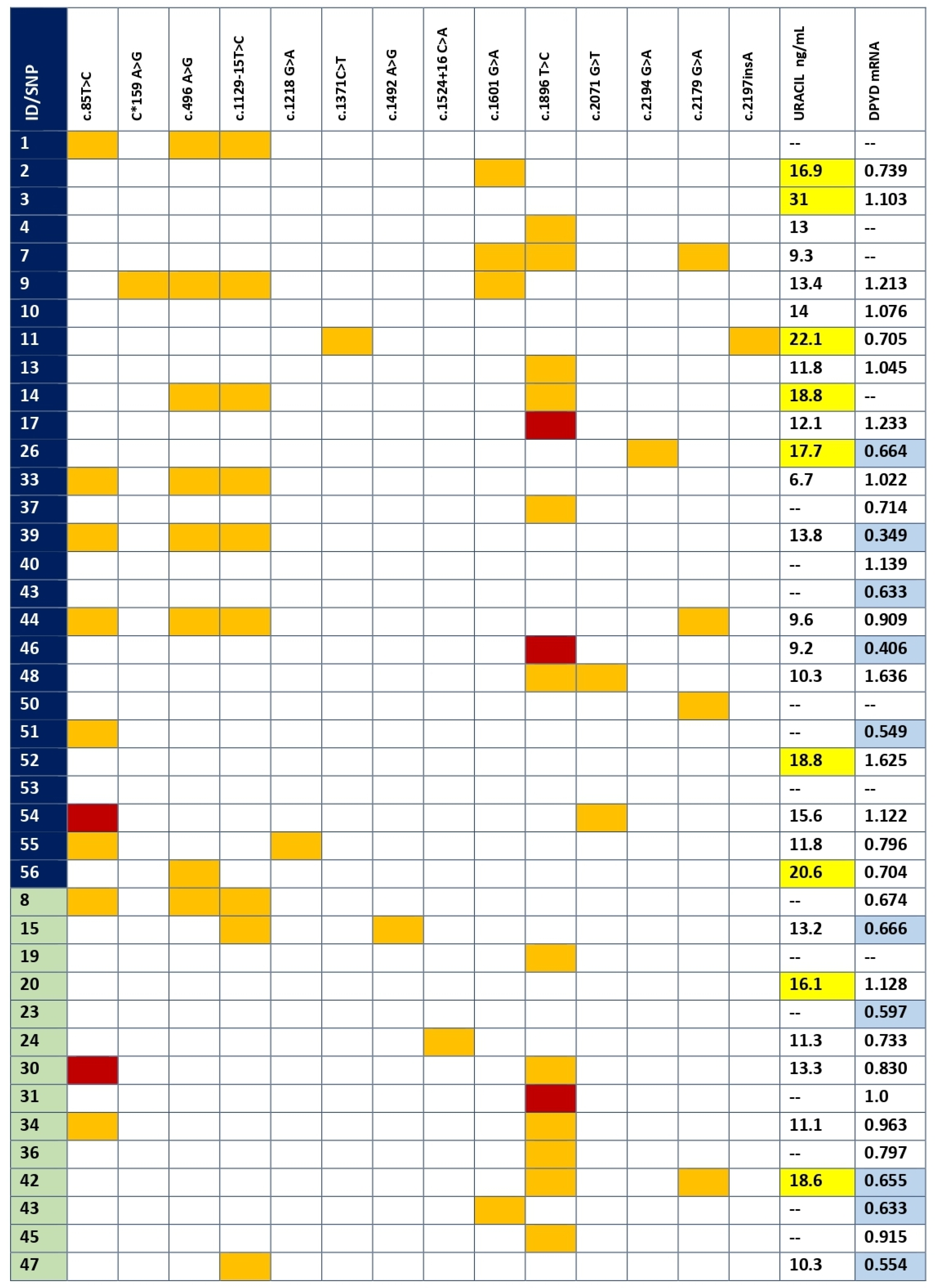
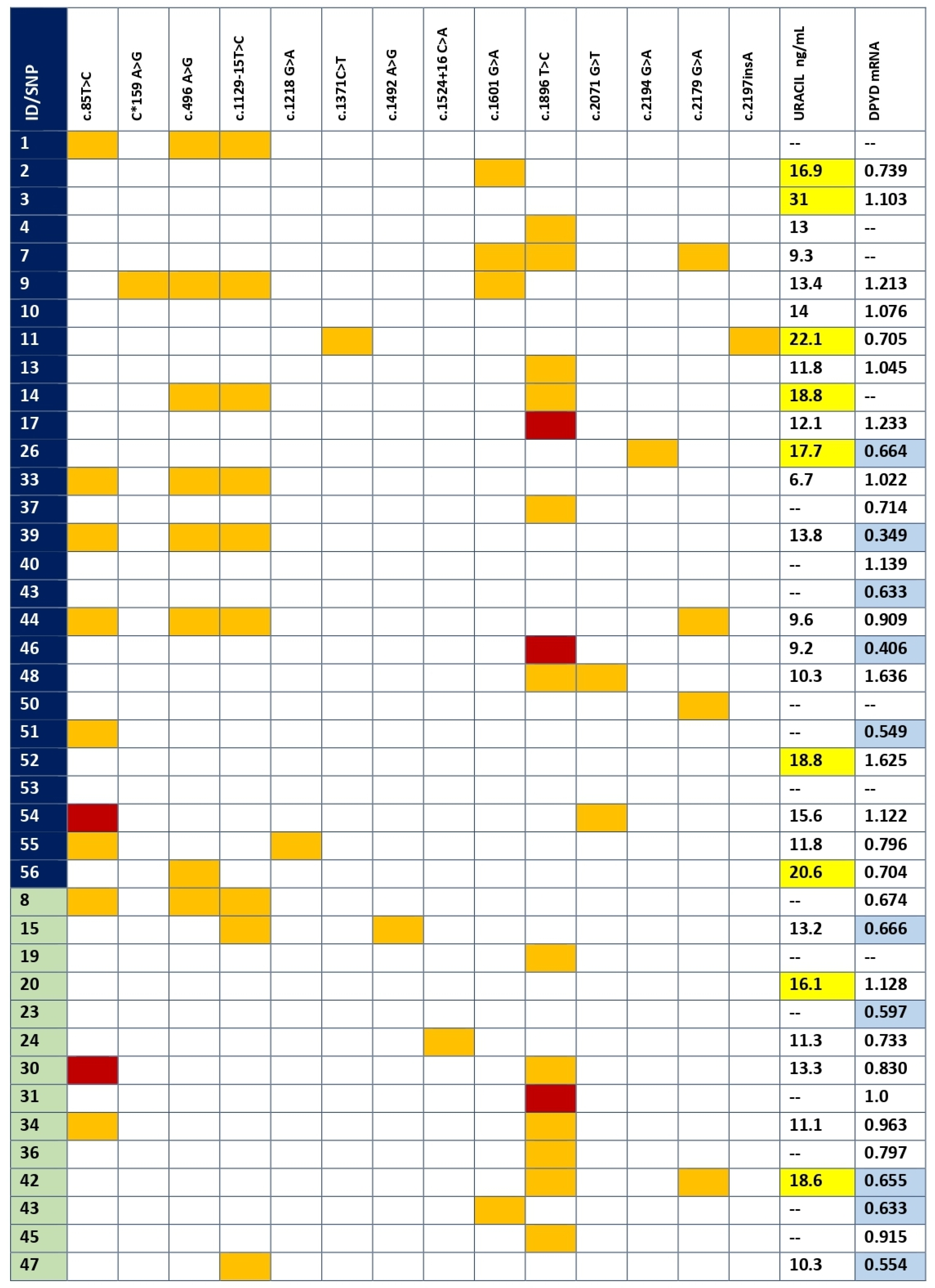
3.2. DPYD Genetic Variants in Coding Regions and Splice Sites
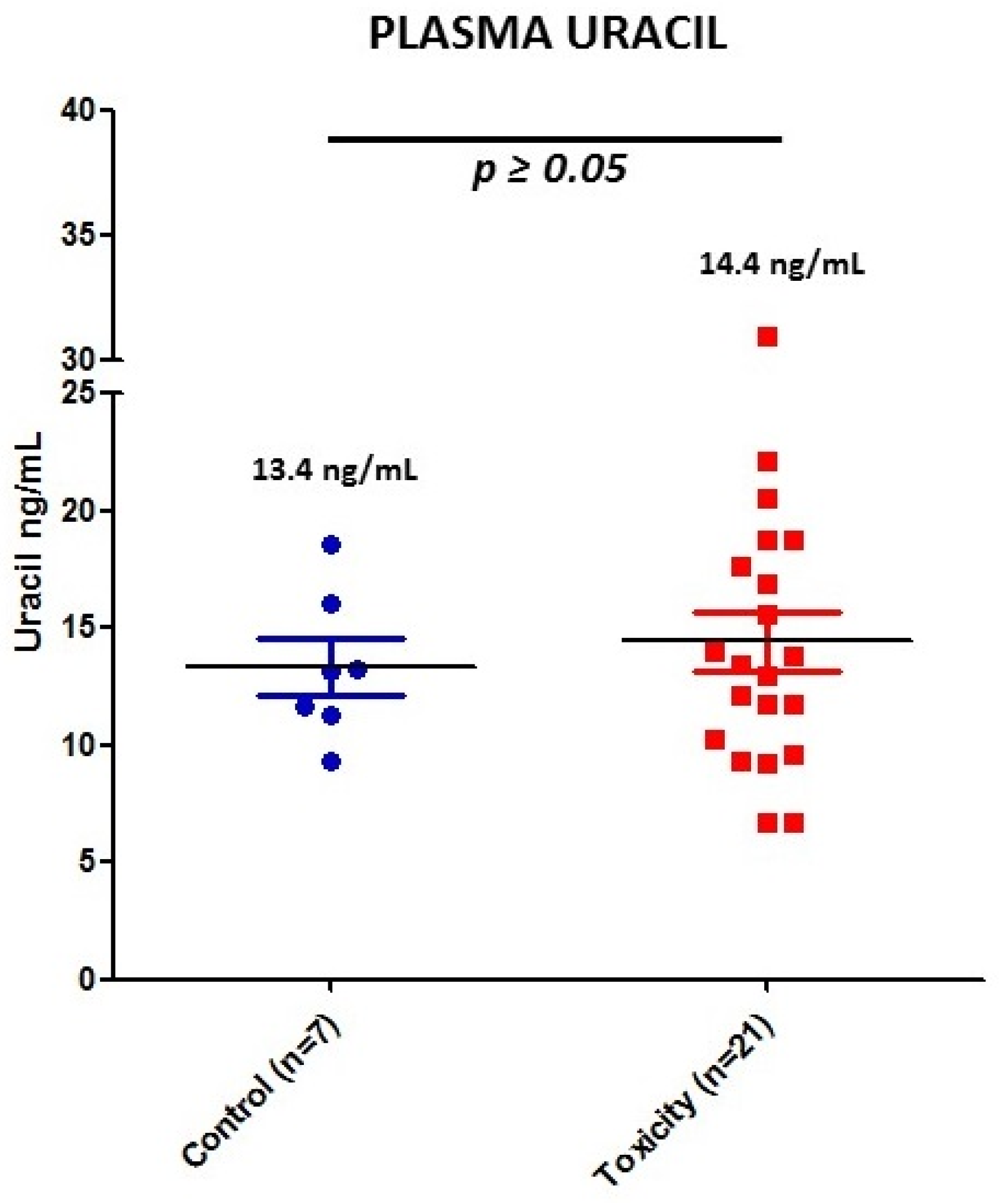
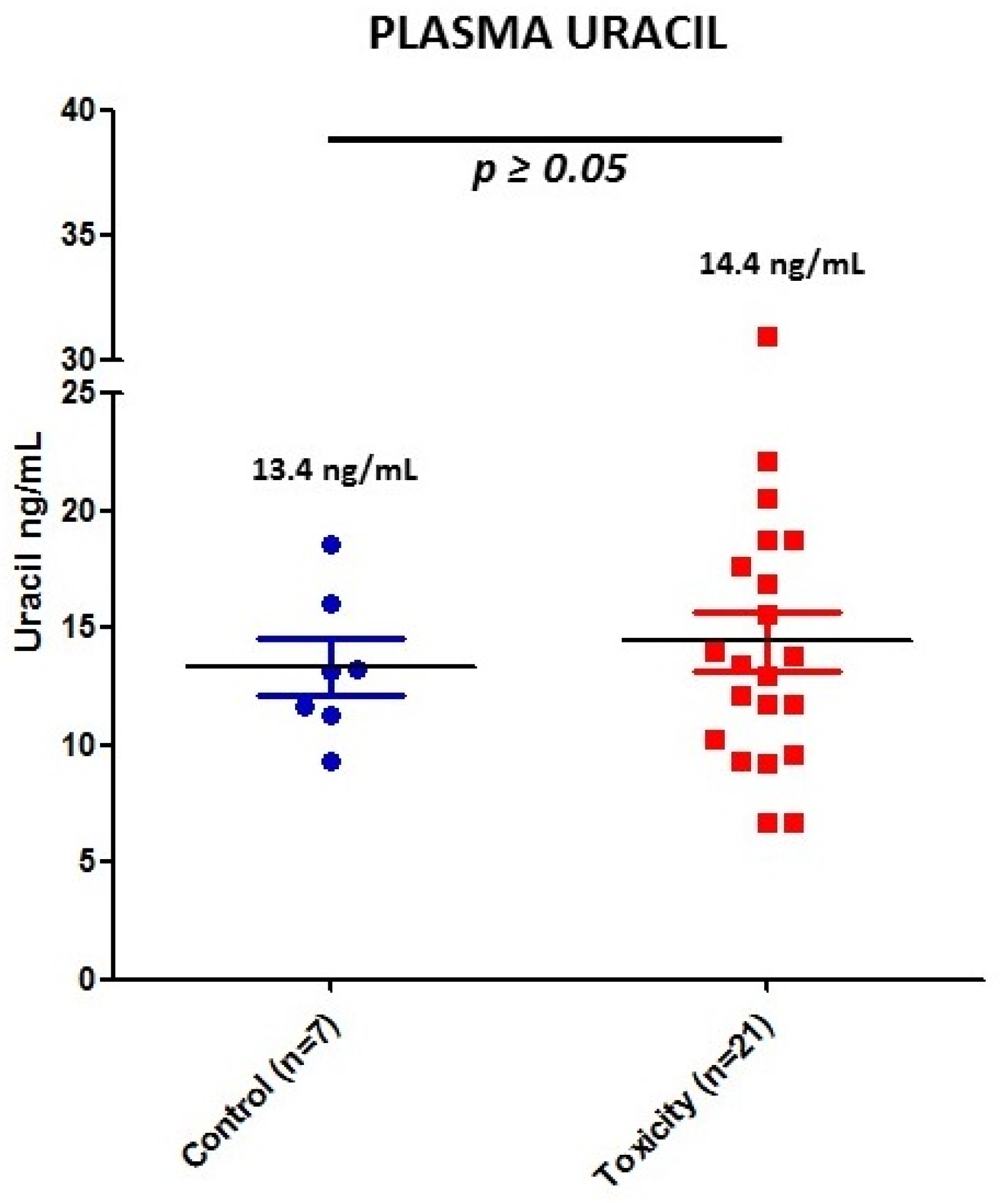
3.3. DPD Activity by Uracil Concentration
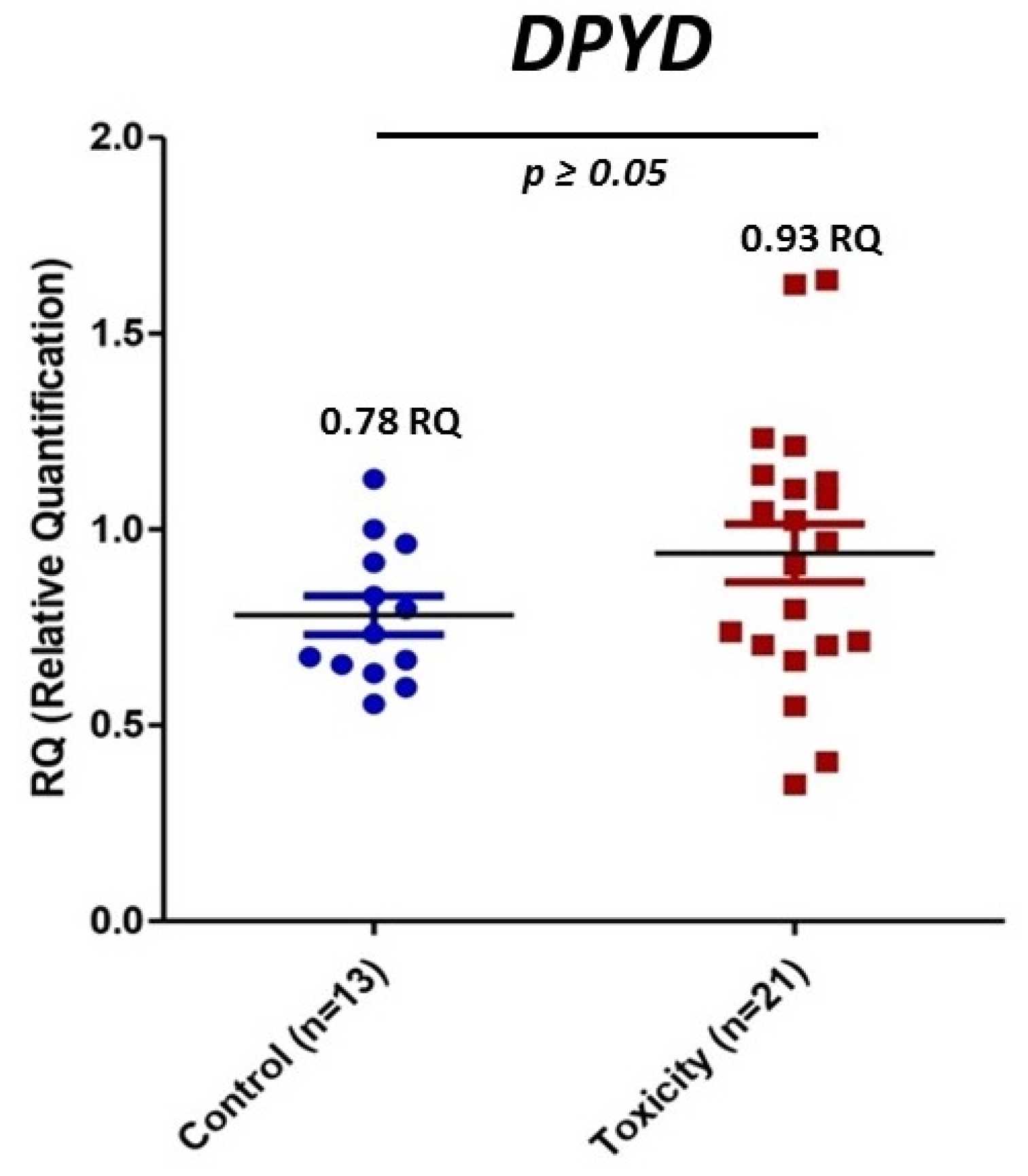
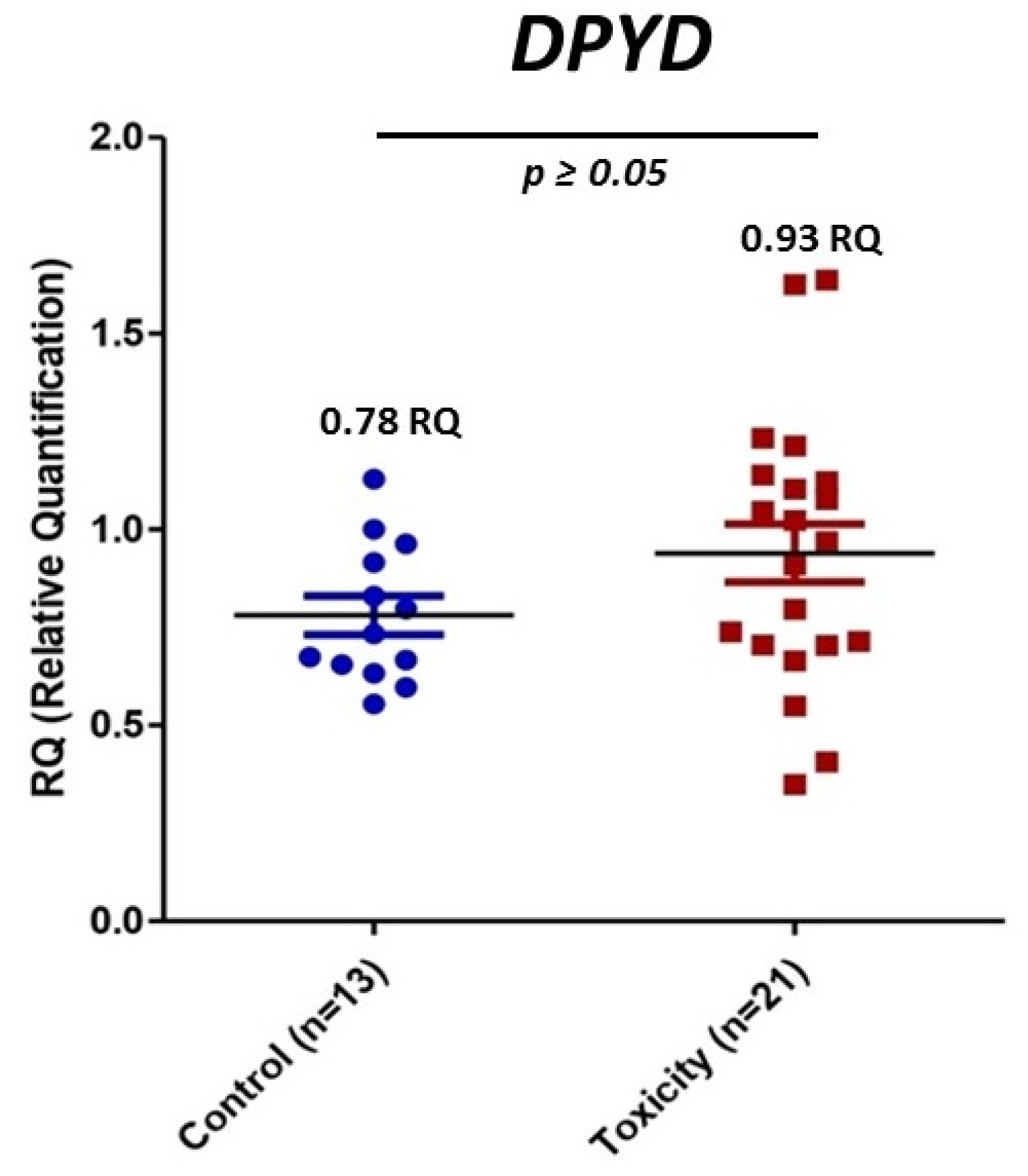
3.4. mRNA DPYD Expression
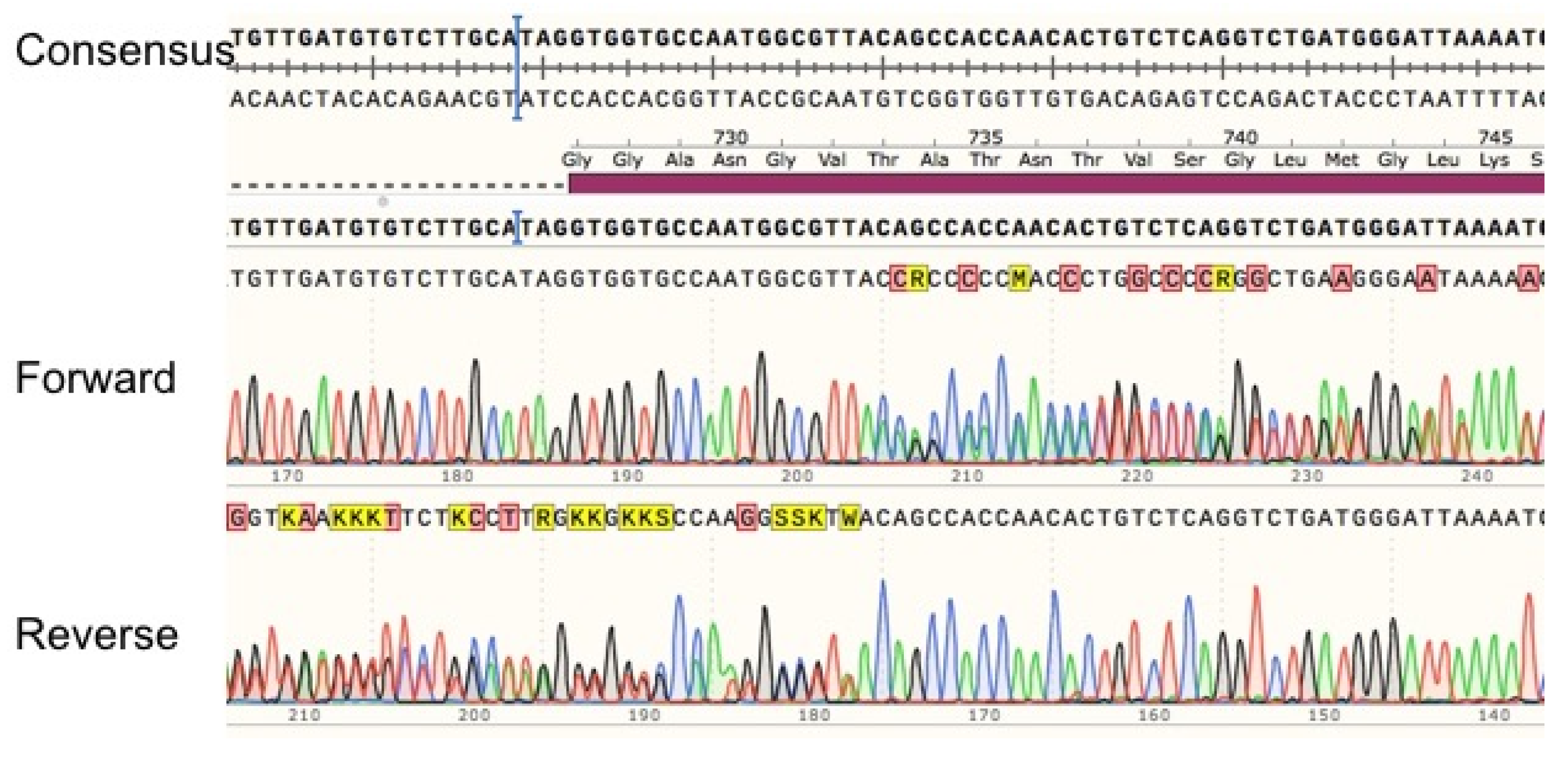
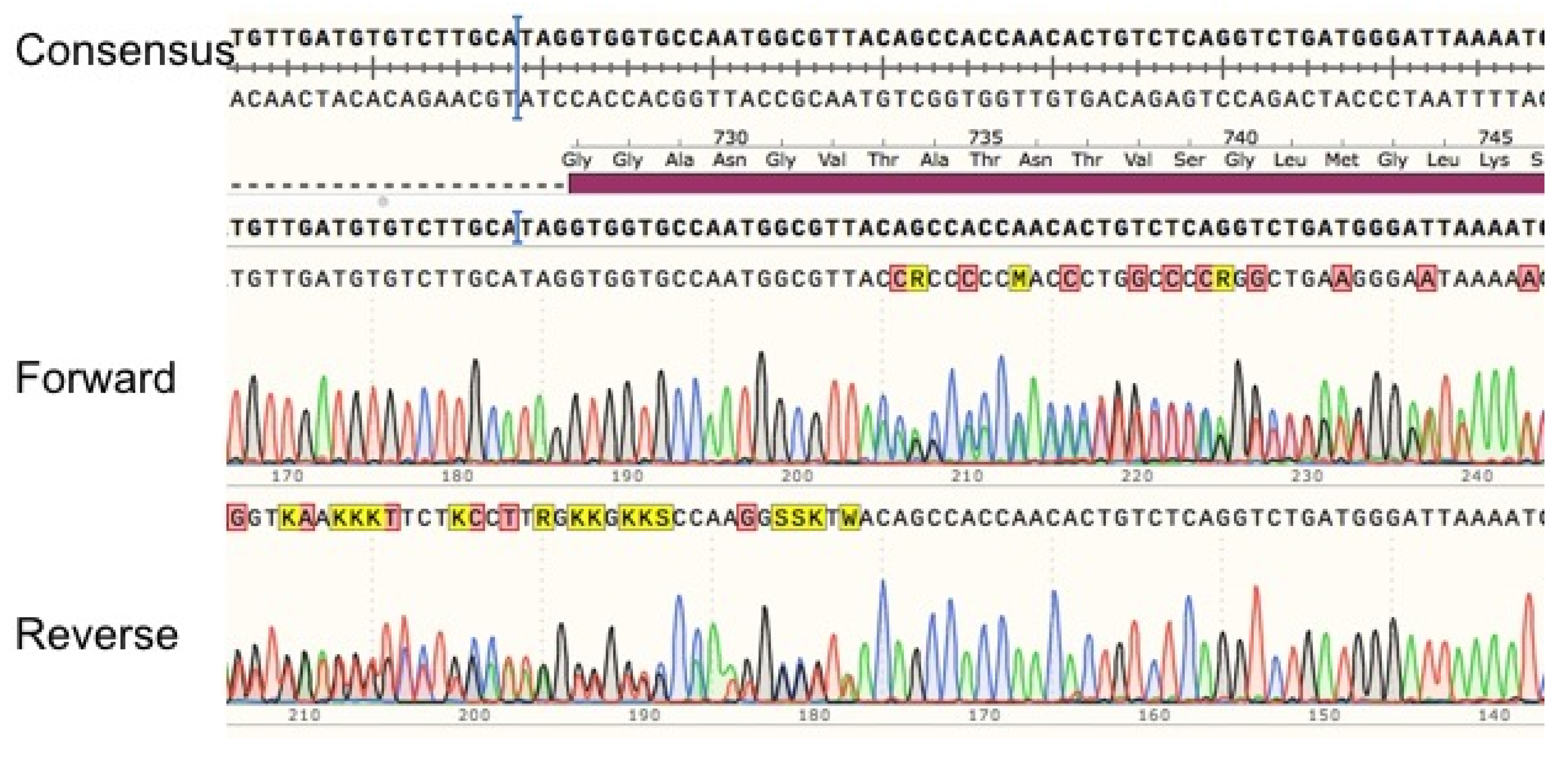
3.5. Characterization of c.2197insA
3.5.1. Sequence Analysis
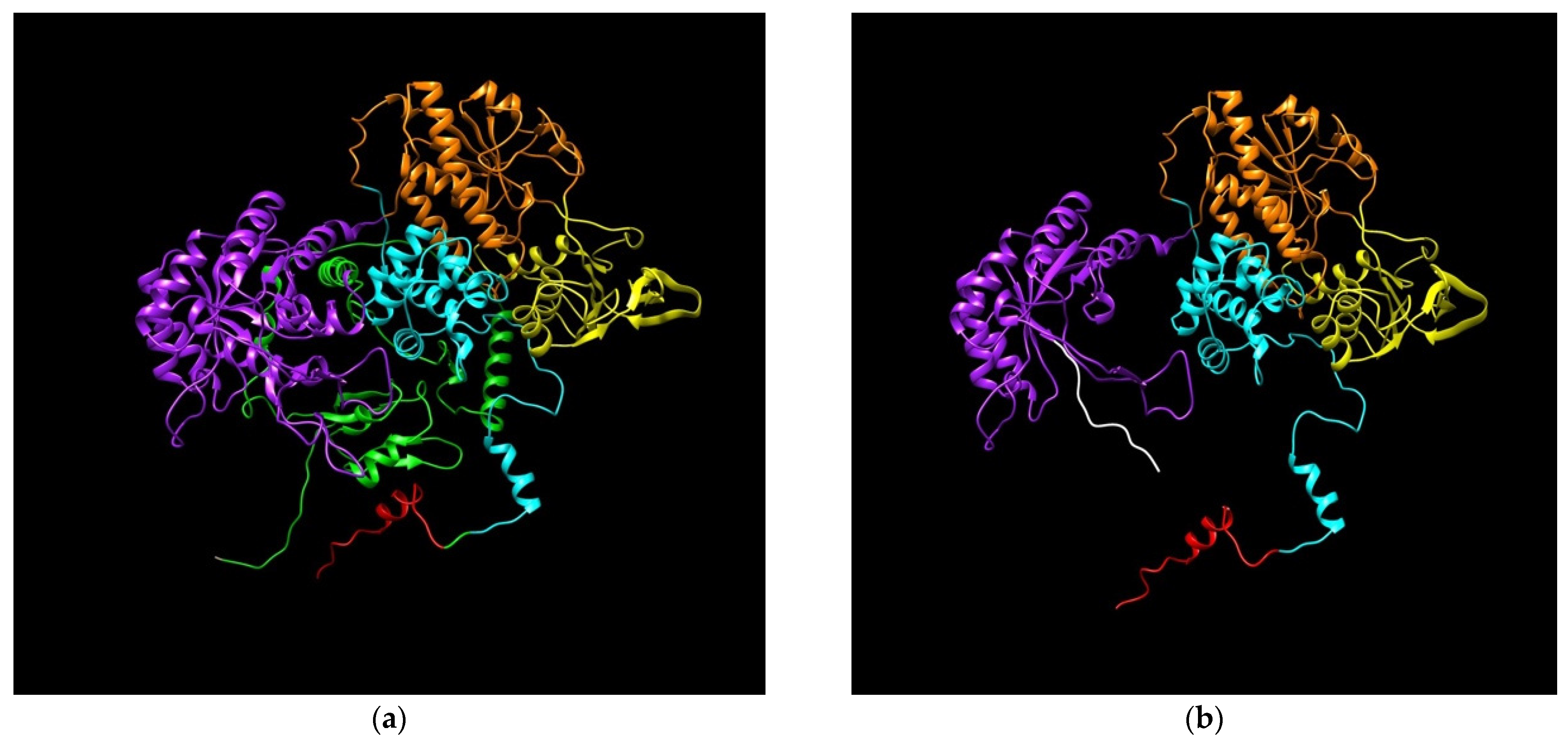
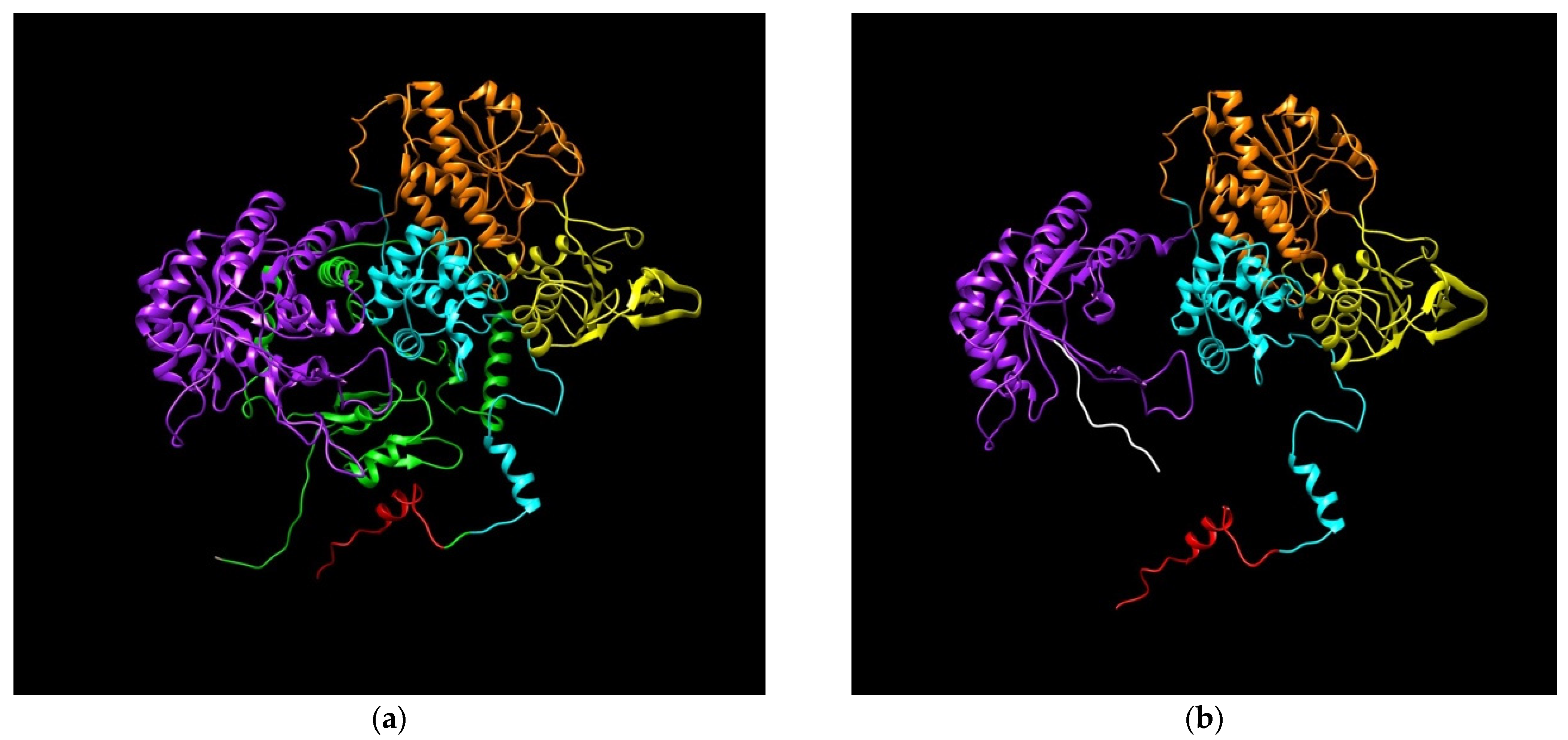
3.5.2. DPD Modeling Generated by c.2197insA Variant
4. Discussion
Future Research Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brito, R.A.; Medgyesy, D.; Zukowski, T.H.; Royce, M.E.; Ravandi-Kashani, F.; Hoff, P.M.; Pazdur, R. Fluoropyrimidines: A critical evaluation. Oncology 1999, 57, 2–8. [Google Scholar] [CrossRef]
- Barin-Le Guellec, C.; Lafay-Chebassier, C.; Ingrand, I.; Tournamille, J.-F.; Boudet, A.; Lanoue, M.-C.; Defossez, G.; Ingrand, P.; Perault-Pochat, M.-C.; Etienne-Grimaldi, M.-C. Toxicities associated with chemotherapy regimens containing a fluoropyrimidine: A real-life evaluation in France. Eur. J. Cancer 2020, 124, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Pallet, N.; Hamdane, S.; Garinet, S.; Blons, H.; Zaanan, A.; Paillaud, E.; Taieb, J.; Laprevote, O.; Loriot, M.-A.; Narjoz, C. A comprehensive population-based study comparing the phenotype and genotype in a pretherapeutic screen of dihydropyrimidine dehydrogenase deficiency. Br. J. Cancer 2020, 123, 811–818. [Google Scholar] [CrossRef]
- Loriot, M.-A.; Ciccolini, J.; Thomas, F.; Barin-Le-Guellec, C.; Royer, B.; Milano, G.; Picard, N.; Becquemont, L.; Verstuyft, C.; Narjoz, C.; et al. Dihydropyrimidine déhydrogenase (DPD) deficiency screening and securing of fluoropyrimidine-based chemotherapies: Update and recommendations of the French GPCO-Unicancer and RNPGx networks. Bull. Cancer 2018, 105, 397–407. [Google Scholar] [CrossRef]
- Katona, C.; Kralovánszky, J.; Rosta, A.; Pandi, E.; Fónyad, G.; Tóth, K.; Jeney, A. Putative role of dihydropyrimidine dehydrogenase in the toxic side effect of 5-fluorouracil in colorectal cancer patients. Oncology 1998, 55, 468–474. [Google Scholar] [CrossRef]
- Stein, B.N.; Petrelli, N.J.; Douglass, H.O.; Driscoll, D.L.; Arcangeli, G.; Meropol, N.J. Age and sex are independent predictors of 5-fluorouracil toxicity. Analysis of a large scale phase III trial. Cancer 1995, 75, 11–17. [Google Scholar] [CrossRef]
- Wagner, A.D.; Grothey, A.; Andre, T.; Dixon, J.G.; Wolmark, N.; Haller, D.G.; Allegra, C.J.; de Gramont, A.; VanCutsem, E.; Alberts, S.R.; et al. Sex and Adverse Events of Adjuvant Chemotherapy in Colon Cancer: An Analysis of 34 640 Patients in the ACCENT Database. J. Natl. Cancer Inst. 2021, 113, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, A.; Graziano, F.; Galli, F.; Galli, F.; Rulli, E.; Lonardi, S.; Ronzoni, M.; Massidda, B.; Zagonel, V.; Pella, N.; et al. Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine-related toxicity in the randomised, phase III adjuvant TOSCA trial in high-risk colon cancer patients. Br. J. Cancer 2017, 117, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Agency, E.M. European Medicines Agency—Multidisciplinary—Multidisciplinary: Pharmacogenomics. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000411.jsp&mid=WC0b01ac058002958e (accessed on 5 June 2015).
- Shakeel, F.; Fang, F.; Kwon, J.W.; Koo, K.; Pasternak, A.L.; Henry, N.L.; Sahai, V.; Kidwell, K.M.; Hertz, D.L. Patients carrying DPYD variant alleles have increased risk of severe toxicity and related treatment modifications during fluoropyrimidine chemotherapy. Pharmacogenomics 2021, 22, 145–155. [Google Scholar] [CrossRef]
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.M.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Guío, A.; Bernabéu-Martínez, Á.; Corno-Caparrós, A.; Aznar-Saliente, T.; Bonete-Sánchez, M.; Calleja-Hernández, M.Á. DPYD variant testing in candidates for fluoropyrimidine treatment: A study protocol. Farm. Hosp. 2021, 45, 155–159. [Google Scholar]
- Collie-Duguid, E.S.; Etienne, M.C.; Milano, G.; McLeod, H.L. Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics 2000, 10, 217–223. [Google Scholar] [CrossRef]
- García-González, X.; Kaczmarczyk, B.; Abarca-Zabalía, J.; Thomas, F.; García-Alfonso, P.; Robles, L.; Pachón, V.; Vaz, Á.; Salvador-Martín, S.; Sanjurjo-Sáez, M.; et al. New DPYD variants causing DPD deficiency in patients treated with fluoropyrimidine. Cancer Chemother. Pharmacol. 2020, 86, 45–54. [Google Scholar] [CrossRef] [PubMed]
- García-González, X.; López-Tarruella, S.; García, M.I.; González-Haba, E.; Blanco, C.; Salvador-Martin, S.; Jerez, Y.; Thomas, F.; Jarama, M.; Sanjurjo Sáez, M.; et al. Severe toxicity to capecitabine due to a new variant at a donor splicing site in the dihydropyrimidine dehydrogenase (DPYD) gene. Cancer Manag. Res. 2018, 10, 4517–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saarenheimo, J.; Wahid, N.; Eigeliene, N.; Ravi, R.; Salomons, G.S.; Ojeda, M.F.; Vijzelaar, R.; Jekunen, A.; van Kuilenburg, A.B.P. Preemptive screening of DPYD as part of clinical practice: High prevalence of a novel exon 4 deletion in the Finnish population. Cancer Chemother. Pharmacol. 2021, 87, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Agency, E.M. EMA Recommendations on DPD Testing Prior to Treatment with Fluorouracil, Capecitabine, Tegafur and Flucytosine. Available online: https://www.ema.europa.eu/en/documents/press-release/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine_en.pdf (accessed on 12 May 2021).
- Spanish Medicine and Health Products Agency (AEMPS) Recommendation on DPD Testing. Available online: https://www.aemps.gob.es/informa/notasinformativas/medicamentosusohumano-3/seguridad-1/2020-seguridad-1/fluorouracilo-capecitabina-tegafur-y-flucitosina-en-pacientes-con-deficit-de-dihidropirimidina-deshidrogenasa/?lang=en (accessed on 12 May 2021).
- Zhang, X.; Soong, R.; Wang, K.; Li, L.; Davie, J.R.; Guarcello, V.; Diasio, R.B. Suppression of DPYD expression in RKO cells via DNA methylation in the regulatory region of the DPYD promoter: A potentially important epigenetic mechanism regulating DPYD expression. Biochem. Cell Biol. 2007, 85, 337–346. [Google Scholar] [CrossRef]
- Dong, S.-Q.; Wang, T.-M.; Zhang, J.-B.; He, Y.-Q.; Xue, W.-Q.; Wu, Z.-Y.; Yang, D.-W.; Cao, L.-J.; Huang, J.-W.; Li, X.-Z.; et al. Polymorphisms in TYMS for Prediction of Capecitabine-Induced Hand-Foot Syndrome in Chinese Patients with Colorectal Cancer. Cancer Res. Treat. 2021, 53, 724–732. [Google Scholar] [CrossRef]
- Ruiz-Pinto, S.; Pita, G.; Martín, M.; Nuñez-Torres, R.; Cuadrado, A.; Shahbazi, M.N.; Caronia, D.; Kojic, A.; Moreno, L.T.; de la Torre-Montero, J.C.; et al. Regulatory CDH4 Genetic Variants Associate With Risk to Develop Capecitabine-Induced Hand-Foot Syndrome. Clin. Pharmacol. Ther. 2021, 109, 462–470. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. LDmatrix tool. Available online: https://ldlink.nci.nih.gov/?tab=ldmatrix (accessed on 12 August 2021).
- Källberg, M.; Margaryan, G.; Wang, S.; Ma, J.; Xu, J. RaptorX server: A resource for template-based protein structure modeling. Methods Mol. Biol. 2014, 1137, 17–27. [Google Scholar] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Deenen, M.J.; Tol, J.; Burylo, A.M.; Doodeman, V.D.; de Boer, A.; Vincent, A.; Guchelaar, H.-J.; Smits, P.H.M.; Beijnen, J.H.; Punt, C.J.A.; et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin. Cancer Res. 2011, 17, 3455–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne-Grimaldi, M.C.; Boyer, J.C.; Beroud, C.; Mbatchi, L.; Van Kuilenburg, A.; Bobin-Dubigeon, C.; Thomas, F.; Chatelut, E.; Merlin, J.L.; Pinguet, F.; et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS ONE 2017, 12, e0175998. [Google Scholar] [CrossRef] [PubMed]
- Dobritzsch, D.; Schneider, G.; Schnackerz, K.D.; Lindqvist, Y. Crystal structure of dihydropyrimidine dehydrogenase, a major determinant of the pharmacokinetics of the anti-cancer drug 5-fluorouracil. EMBO J. 2001, 20, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Shulman, E.D.; Elkon, R. Systematic identification of functional SNPs interrupting 3’UTR polyadenylation signals. PLoS Genet. 2020, 16, e1008977. [Google Scholar] [CrossRef]
- Pamuła-Piłat, J.; Tęcza, K.; Kalinowska-Herok, M.; Grzybowska, E. Genetic 3’UTR variations and clinical factors significantly contribute to survival prediction and clinical response in breast cancer patients. Sci. Rep. 2020, 10, 5736. [Google Scholar] [CrossRef]
- Offer, S.M.; Fossum, C.C.; Wegner, N.J.; Stuflesser, A.J.; Butterfield, G.L.; Diasio, R.B. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014, 74, 2545–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, S.; Zhang, C.; Jerde, C.R.; Nie, Q.; Li, H.; Offer, S.M.; Diasio, R.B. Gene-Specific Variant Classifier (DPYD-Varifier) to Identify Deleterious Alleles of Dihydropyrimidine Dehydrogenase. Clin. Pharmacol. Ther. 2018, 104, 709–718. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; van der Wouden, C.H.; Nijenhuis, M.; Crommentuijn-van Rhenen, M.H.; de Boer-Veger, N.J.; Buunk, A.M.; Houwink, E.J.F.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. 2020, 28, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, G.; Giodini, L.; Buonadonna, A.; Berretta, M.; De Paoli, A.; Scalone, S.; Miolo, G.; Mini, E.; Nobili, S.; Lonardi, S.; et al. Clinical validity of a DPYD-based pharmacogenetic test to predict severe toxicity to fluoropyrimidines. Int. J. Cancer 2015, 137, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Zanger, U.M.; Marx, C.; Schaeffeler, E.; Klein, K.; Dippon, J.; Kerb, R.; Blievernicht, J.; Fischer, J.; Hofmann, U.; et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: A prospective clinical trial by the German 5-FU toxicity study group. J. Clin. Oncol. 2008, 26, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, M.; García-González, X.; García, M.I.; Blanco, C.; García-Alfonso, P.; Robles, L.; Grávalos, C.; Rueda, D.; Martínez, J.; Pachón, V.; et al. Use of exome sequencing to determine the full profile of genetic variants in the fluoropyrimidine pathway in colorectal cancer patients affected by severe toxicity. Pharmacogenomics 2017, 18, 1215–1223. [Google Scholar] [CrossRef]
- Hariprakash, J.M.; Vellarikkal, S.K.; Keechilat, P.; Verma, A.; Jayarajan, R.; Dixit, V.; Ravi, R.; Senthivel, V.; Kumar, A.; Sehgal, P.; et al. Pharmacogenetic landscape of DPYD variants in south Asian populations by integration of genome-scale data. Pharmacogenomics 2018, 19, 227–241. [Google Scholar] [CrossRef]
- Kleibl, Z.; Fidlerova, J.; Kleiblova, P.; Kormunda, S.; Bilek, M.; Bouskova, K.; Sevcik, J.; Novotny, J. Influence of dihydropyrimidine dehydrogenase gene (DPYD) coding sequence variants on the development of fluoropyrimidine-related toxicity in patients with high-grade toxicity and patients with excellent tolerance of fluoropyrimidine-based chemotherapy. Neoplasma 2009, 56, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.; Busse, B.; Riemenschneider, M.; Neubauer, S.; Seck, K.; Klein, H.-G.; Kiechle, M.; Lordick, F.; Meindl, A. Strong association of a common dihydropyrimidine dehydrogenase gene polymorphism with fluoropyrimidine-related toxicity in cancer patients. PLoS ONE 2008, 3, e4003. [Google Scholar] [CrossRef] [Green Version]
- Del Re, M.; Cinieri, S.; Michelucci, A.; Salvadori, S.; Loupakis, F.; Schirripa, M.; Cremolini, C.; Crucitta, S.; Barbara, C.; Di Leo, A.; et al. DPYD*6 plays an important role in fluoropyrimidine toxicity in addition to DPYD*2A and c.2846A>T: A comprehensive analysis in 1254 patients. Pharm. J. 2019, 19, 556–563. [Google Scholar] [CrossRef]
- Falvella, F.S.; Cheli, S.; Martinetti, A.; Mazzali, C.; Iacovelli, R.; Maggi, C.; Gariboldi, M.; Pierotti, M.A.; Di Bartolomeo, M.; Sottotetti, E.; et al. DPD and UGT1A1 deficiency in colorectal cancer patients receiving triplet chemotherapy with fluoropyrimidines, oxaliplatin and irinotecan. Br. J. Clin. Pharmacol. 2015, 80, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Hamzic, S.; Schärer, D.; Offer, S.M.; Meulendijks, D.; Nakas, C.; Diasio, R.B.; Fontana, S.; Wehrli, M.; Schürch, S.; Amstutz, U.; et al. Haplotype structure defines effects of common DPYD variants c.85T > C (rs1801265) and c.496A > G (rs2297595) on dihydropyrimidine dehydrogenase activity: Implication for 5-fluorouracil toxicity. Br. J. Clin. Pharmacol. 2021, 87, 3234–3243. [Google Scholar] [CrossRef]
- Gokare, P.; Finnberg, N.K.; Abbosh, P.H.; Dai, J.; Murphy, M.E.; El-Deiry, W.S. P53 represses pyrimidine catabolic gene dihydropyrimidine dehydrogenase (DPYD) expression in response to thymidylate synthase (TS) targeting. Sci. Rep. 2017, 7, 9711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Zhang, R.; Diasio, R.B. Population characteristics of hepatic dihydropyrimidine dehydrogenase activity, a key metabolic enzyme in 5-fluorouracil chemotherapy. Clin. Pharmacol. Ther. 1995, 58, 512–522. [Google Scholar] [CrossRef]
- Uetake, H.; Ichikawa, W.; Takechi, T.; Fukushima, M.; Nihei, Z.; Sugihara, K. Relationship between intratumoral dihydropyrimidine dehydrogenase activity and gene expression in human colorectal cancer. Clin. Cancer Res. 1999, 5, 2836–2839. [Google Scholar]
- Wigle, T.J.; Tsvetkova, E.V.; Welch, S.A.; Kim, R.B. DPYD and Fluorouracil-Based Chemotherapy: Mini Review and Case Report. Pharmaceutics 2019, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Naiki-Ito, A.; Suzuki, S.; Inaguma, S.; Komura, M.; Nakao, K.; Naiki, T.; Kachi, K.; Kato, A.; Matsuo, Y.; et al. DPYD, down-regulated by the potentially chemopreventive agent luteolin, interacts with STAT3 in pancreatic cancer. Carcinogenesis 2021, 42, 940–950. [Google Scholar] [CrossRef]
- Milano, G.; Fischel, J.L.; Etienne, M.C.; Renée, N.; Formento, P.; Thyss, A.; Gaspard, M.H.; Thill, L.; Cupissol, D. Inhibition of dihydropyrimidine dehydrogenase by alpha-interferon: Experimental data on human tumor cell lines. Cancer Chemother. Pharmacol. 1994, 34, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Sakowicz-Burkiewicz, M.; Przybyla, T.; Wesserling, M.; Bielarczyk, H.; Maciejewska, I.; Pawelczyk, T. Suppression of TWIST1 enhances the sensitivity of colon cancer cells to 5-fluorouracil. Int. J. Biochem. Cell Biol. 2016, 78, 268–278. [Google Scholar] [CrossRef]
- Lin, Y.; Gao, Z.-X.; Shen, X.; Chen, M.-J.; Li, Y.-T.; Li, S.-L.; Lin, H.-L.; Zhao, Q.-F.; Liu, F.; Niu, J.-J. Correlation between polymorphisms in toll-like receptor genes and the activity of hepatitis B virus among treatment-naïve patients: A case-control study in a Han Chinese population. BMC Infect. Dis. 2018, 18, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offer, S.M.; Butterfield, G.L.; Jerde, C.R.; Fossum, C.C.; Wegner, N.J.; Diasio, R.B. MicroRNAs miR-27a and miR-27b directly regulate liver dihydropyrimidine dehydrogenase expression through two conserved binding sites. Mol. Cancer Ther. 2014, 13, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Relevance of DNA methylation in the management of cancer. Lancet. Oncol. 2003, 4, 351–358. [Google Scholar] [CrossRef]
- Ezzeldin, H.H.; Lee, A.M.; Mattison, L.K.; Diasio, R.B. Methylation of the DPYD promoter: An alternative mechanism for dihydropyrimidine dehydrogenase deficiency in cancer patients. Clin. Cancer Res. 2005, 11, 8699–8705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavani, O. 5-Fluorouracil Response Prediction and Blood Level-Guided Therapy in Oncology: Existing Evidence Fundamentally Supports Instigation. Ther. Drug Monit. 2020, 42, 660–664. [Google Scholar] [CrossRef]
- Thomas, F.; Maillard, M.; Launay, M.; Tron, C.; Etienne-Grimaldi, M.-C.; Gautier-Veyret, E.; Haufroid, V.; Pallet, N.; Royer, B.; Narjoz, C.; et al. Artificial increase of uracilemia during fluoropyrimidine treatment can lead to DPD deficiency misinterpretation. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 810–811. [Google Scholar] [CrossRef] [PubMed]
- Knikman, J.E.; Gelderblom, H.; Beijnen, J.H.; Cats, A.; Guchelaar, H.-J.; Henricks, L.M. Individualized Dosing of Fluoropyrimidine-Based Chemotherapy to Prevent Severe Fluoropyrimidine-Related Toxicity: What Are the Options? Clin. Pharmacol. Ther. 2021, 109, 591–604. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Overall (n = 41) | Toxicity Group (n = 27) | Control Group (n = 14) | p Value |
|---|---|---|---|---|
| Age at diagnosis (IQR, range; years) | 63 (17, 36–81) | 63 (16, 36–81) | 63 (19, 36–80) | 0.805 |
| Gender | ||||
| Male, n (%) | 20 | 12 (42.9%) | 8 (57.1%) | 0.382 |
| Female, n (%) | 21 | 15 (55.5%) | 6 (42.9%) | |
| Type of cancer | ||||
| Gastric | 7 | 5 (18.5%) | 2 (14.3%) | 0.821 |
| Rectum | 7 | 4 (14.8%) | 3 (21.4%) | |
| Colon | 21 | 13 (48.1%) | 8 (57.1%) | |
| Breast | 2 | 2 (7.4%) | 0 (0.0%) | |
| Pancreas | 4 | 3 (11.1%) | 1 (7.1%) | |
| Clinical stage | ||||
| I | 0 | 0 (0.0%) | 0 (0.0%) | 0.347 |
| IIA | 2 | 2 (7.4%) | 0 (0.0%) | |
| IIB | 2 | 1 (3.7%) | 1 (7.1%) | |
| IIIA | 1 | 1 (3.7%) | 0 (0.0%) | |
| IIIB | 10 | 9 (33.3%) | 1 (7.1%) | |
| IIIC | 5 | 3 (11.1%) | 2 (14.3%) | |
| IV | 21 | 11 (40.7%) | 10 (71.4%) | |
| Surgery, yes n (%) | 33 | 23 (85.1%) | 10 (71.4%) | 0.292 |
| Complete resection, yes n (%) | 26 | 18 (66.6%) | 8 (57.1%) | 0.489 |
| Neoadjuvant treatment, yes n (%) | 7 | 4 (14.8%) | 3 (21.4%) | 0.321 |
| Type of chemotherapy | ||||
| 5-fluorouracil | 16 | 14 (51.9%) | 2 (14.3%) | 0.025 * |
| Capecitabine | 25 | 13 (48.1%) | 12 (85.7%) | |
| Accumulated dose (g) | ||||
| 5-fluorouracil | 3.4 (5.0, 2.3–13.6) | 3.3 (2.3, 2.3–13.6) | 10.0 (NA, 9.0–11.1) | 0.047 * |
| Capecitabine | 13.1 (8.7, 2.2–16.6) | 7.7 (10.3, 2.5–16.6) | 13.9 (3.9, 2.2–16.6) | 0.031 * |
| Concomitant medication | ||||
| Oxaliplatin | 31 | 20 (74.0%) | 11 (78.6%) | 0.620 |
| Irinotecan | 4 | 2 (7.4%) | 2 (14.3%) | 0.457 |
| Cetuximab | 1 | 0 (0.0%) | 1 (7.1%) | 0.152 |
| Panitumumab | 2 | 1 (3.7%) | 1 (7.1%) | 0.608 |
| Bevacizumab | 5 | 2 (7.4%) | 3 (21.4%) | 0.178 |
| Trastuzumab | 2 | 1 (3.6%) | 1 (7.1%) | 0.608 |
| Complete cycles | ||||
| 1 | 41 | 27 (100%) | 14 (100%) | NA |
| 2 | 27 | 13 (48.1%) | 14 (100%) | 0.001 * |
| 3 | 20 | 6 (22.2%) | 14 (100%) | 0.000002 * |
| Dose reduction due to toxicity | ||||
| Cycle 1 | 4 | 3 (11.1%) | 1(7.1%) | 0.709 |
| Cycle 2 | 6 | 6 (40%) | 0 (0.0%) | 0.039 * |
| Cycle 3 | 7 | 6 (100%) | 1 (7.1%) | 0.002 * |
| Adverse Event | Overall (n = 41) | Toxicity Group (n = 27) | Control Group (n = 14) | p Value |
|---|---|---|---|---|
| Hemoglobin | ||||
| Hemoglobin, g/dL # | 11.9 (1.2, 7.2–14.7) | 11.2 (3.1, 7.2–14.7) | 12.9 (1.9, 11–13.7) | 0.025 * |
| Anemia, n (%) | 19 (46%) | 15 (55%) | 4 (28%) | 0.125 |
| Severe anemia (≥Grade 3), n (%) | 2 (5%) | 2 (7%) | 0 (0%) | 0.440 |
| Leukocytes | ||||
| Leukocyte count × 103/mm3 # | 4.3 (3.2, 0.3–8.9) | 3.1 (2, 0.3–8.9) | 5.9 (2.1, 3.6–8.7) | 0.003 * |
| Leukopenia, n (%) | 19 (46%) | 17 (63%) | 2 (14%) | 0.004 * |
| Severe leukopenia (≥Grade 3), n (%) | 3 (7%) | 3 (11%) | 0 (0%) | 0.517 |
| Neutrophils | ||||
| Neutrophil count × 103/mm3 # | 1.8 (2.4, 0.1–6.3) | 1.2 (1.7, 0.1–6.3) | 2.8 (1.7, 1.5–5) | 0.009 * |
| Neutropenia, n (%) | 22 (54%) | 20 (74%) | 2 (14%) | <0.001 * |
| Severe neutropenia (≥Grade 3), n (%) | 12 (55%) | 12 (60%) | 0 (0%) | 0.104 |
| Lymphocytes | ||||
| Lymphocyte count ×103/mm3 # | 1.1 (0.8, 0.1–3) | 1.0 (0.7, 0.1–2.6) | 1.8 (1, 0.9–3) | 0.003 * |
| Lymphopenia, n (%) | 21 (51%) | 17 (63%) | 4 (28%) | 0.037 * |
| Severe lymphopenia | 0 | 0 | 0 | NA |
| Platelets | ||||
| Platelet count × 103/mm3 # | 149 (80.2, 14–506) | 148 (94.2, 14–506) | 162.5 (81.2, 102–252) | 0.797 |
| Thrombocitopenia, n (%) | 22 (54%) | 15 (56%) | 7 (50%) | 0.827 |
| Severe thrombocitopenia (≥Grade 3), n (%) | 0 | 0 | 0 | 0 |
| ALT, IU # | 33 (29.5, 7.0–278.0) | 35.5 (56.0, 7.0–278.0) | 30.0 (27.7, 12.0–66.0) | 0.162 |
| Total bilirubin, mg/dL # | 0.5 (0.6, 0.2–2.6) | 0.7 (1, 0.3–2.6) | 0.5 (0.4, 0.2–1.7) | 0.165 |
| GGT, IU | 33 (44, 10–1028) | 33.5 (44, 10–1028) | 47.5 (72.5, 15–365) | 0.971 |
| Creatinine, mg/dL # | 0.8 (0.27, 0.17–1.4) | 0.8 (0.3, 0.1–1.4) | 0.8 (0.1, 0.6–1.1) | 0.917 |
| AP, mg/dL # | 90 (64.0, 44–721) | 92.5 (95.2, 44–721) | 93.5 (42.2, 71–412) | 0.798 |
| Diarrhea, n (%) | 24 (58%) | 21 (78%) | 3 (21%) | 0.008 * |
| Severe diarrhea (≥Grade 3), n (%) | 13 (32%) | 13 (48%) | 0 (0%) | 0.055 |
| Nausea and/or vomiting, n (%) | 18 (43%) | 22 (78%) | 2 (14%) | 0.023 * |
| Severe nausea and/or vomiting, (≥Grade 3), n (%) | 6 (15%) | 6 (22%) | 0 (0%) | 0.209 |
| Mucositis, n (%) | 14 (34%) | 11 (41%) | 2 (14%) | 0.309 |
| Severe mucositis (≥Grade 3), n (%) | 4 (10%) | 4 (15%) | 0 (0%) | 0.305 |
| Hand–foot syndrome, n (%) | 4 (10%) | 4 (15%) | 0 (0%) | 0.331 |
| dbSNP ID | nt Change | aa Change | MAF | ClinVar SIFT PolyPhen-2 | Toxicity n = 27 (%) | Control n = 14 (%) |
|---|---|---|---|---|---|---|
| Toxicity group | ||||||
| rs61622928 | c.1218G>A | p.Met406Ile | 0.0003258 | Controversial | 1 (3.7%) | 0 (0.0%) |
| rs57918000 | c.1371C>T | p.Asn457= | 0.0001321 | Benign | 1 (3.7%) | 0 (0.0%) |
| rs17376848 | c.1896T>C | p.Phe632= | 0.04220 | Benign | 2 (7.4%) | 0 (0.0%) |
| rs202212118 | c.2071G>T | p.Val691Leu | 0.0001167 | Controversial | 1 (3.7%) | 0 (0.0%) |
| c.2197insA | p.Thr733AsnfsTer14 | - | 1 (3.7%) | 0 (0.0%) | ||
| c*159A>G | - | 1 (3.7%) | 0 (0.0%) | |||
| Toxicity and control groups | ||||||
| rs1801265 | c.85T>C | p.Cys29Arg | 0.7755 | Benign | 7 (25.9%) | 3 (21.4%) |
| rs2297595 | c.496A>G | p.Met166Val | 0.1018 | Controversial | 7 (25.9%) | 1 (7.1%) |
| rs56293913 | c.1129-15T>C | NA | 0.1177 | Benign | 6 (22.2%) | 3 (21.4%) |
| rs1801158 | c.1601G>A | p.Ser534Asn | 0.011961 | Controversial | 3 (11.1%) | 1 (7.1%) |
| rs1801159 | c.1627A>G | p.Ile543Val | 0.198899 | Benign | 8 (29.6%) | 7 (50%) |
| rs1801160 | c.2194G>A | p.Val732Ile | 0.04567 | Benign | 3 (11.1%) | 1 (7.1%) |
| Control group | ||||||
| rs116364703 | c.1492A>G | p.Gln498= | 0.00000776 | Benign | 0 (0.0%) | 1 (7.1%) |
| rs199469537 | c.1524+16C>A | NA | 0.0007619 | Benign | 0 (0.0%) | 1 (7.1%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalvazo, P.; Marzal-Alfaro, B.; García-Alfonso, P.; Revuelta-Herrero, J.L.; Thomas, F.; López-Tarruella, S.; García-González, X.; Calvo, A.; Yakoubi, M.; Salvador-Martín, S.; et al. DPYD Exome, mRNA Expression and Uracil Levels in Early Severe Toxicity to Fluoropyrimidines: An Extreme Phenotype Approach. J. Pers. Med. 2021, 11, 792. https://doi.org/10.3390/jpm11080792
Villalvazo P, Marzal-Alfaro B, García-Alfonso P, Revuelta-Herrero JL, Thomas F, López-Tarruella S, García-González X, Calvo A, Yakoubi M, Salvador-Martín S, et al. DPYD Exome, mRNA Expression and Uracil Levels in Early Severe Toxicity to Fluoropyrimidines: An Extreme Phenotype Approach. Journal of Personalized Medicine. 2021; 11(8):792. https://doi.org/10.3390/jpm11080792
Chicago/Turabian StyleVillalvazo, Priscila, Belén Marzal-Alfaro, Pilar García-Alfonso, José Luis Revuelta-Herrero, Fabienne Thomas, Sara López-Tarruella, Xandra García-González, Aitana Calvo, Malika Yakoubi, Sara Salvador-Martín, and et al. 2021. "DPYD Exome, mRNA Expression and Uracil Levels in Early Severe Toxicity to Fluoropyrimidines: An Extreme Phenotype Approach" Journal of Personalized Medicine 11, no. 8: 792. https://doi.org/10.3390/jpm11080792
APA StyleVillalvazo, P., Marzal-Alfaro, B., García-Alfonso, P., Revuelta-Herrero, J. L., Thomas, F., López-Tarruella, S., García-González, X., Calvo, A., Yakoubi, M., Salvador-Martín, S., López-López, F., Aguilar, I., Sanjurjo-Sáez, M., Martín, M., & López-Fernández, L. A. (2021). DPYD Exome, mRNA Expression and Uracil Levels in Early Severe Toxicity to Fluoropyrimidines: An Extreme Phenotype Approach. Journal of Personalized Medicine, 11(8), 792. https://doi.org/10.3390/jpm11080792