
Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis


Abstract
1. Introduction
2. Principle of Intestinal Current Measurement (ICM)
3. ICM as a Biomarker of CFTR Function and Diagnostic Test for CF
4. ICM as Outcome Measure of In Vivo Response to CFTR-Directed Therapeutics
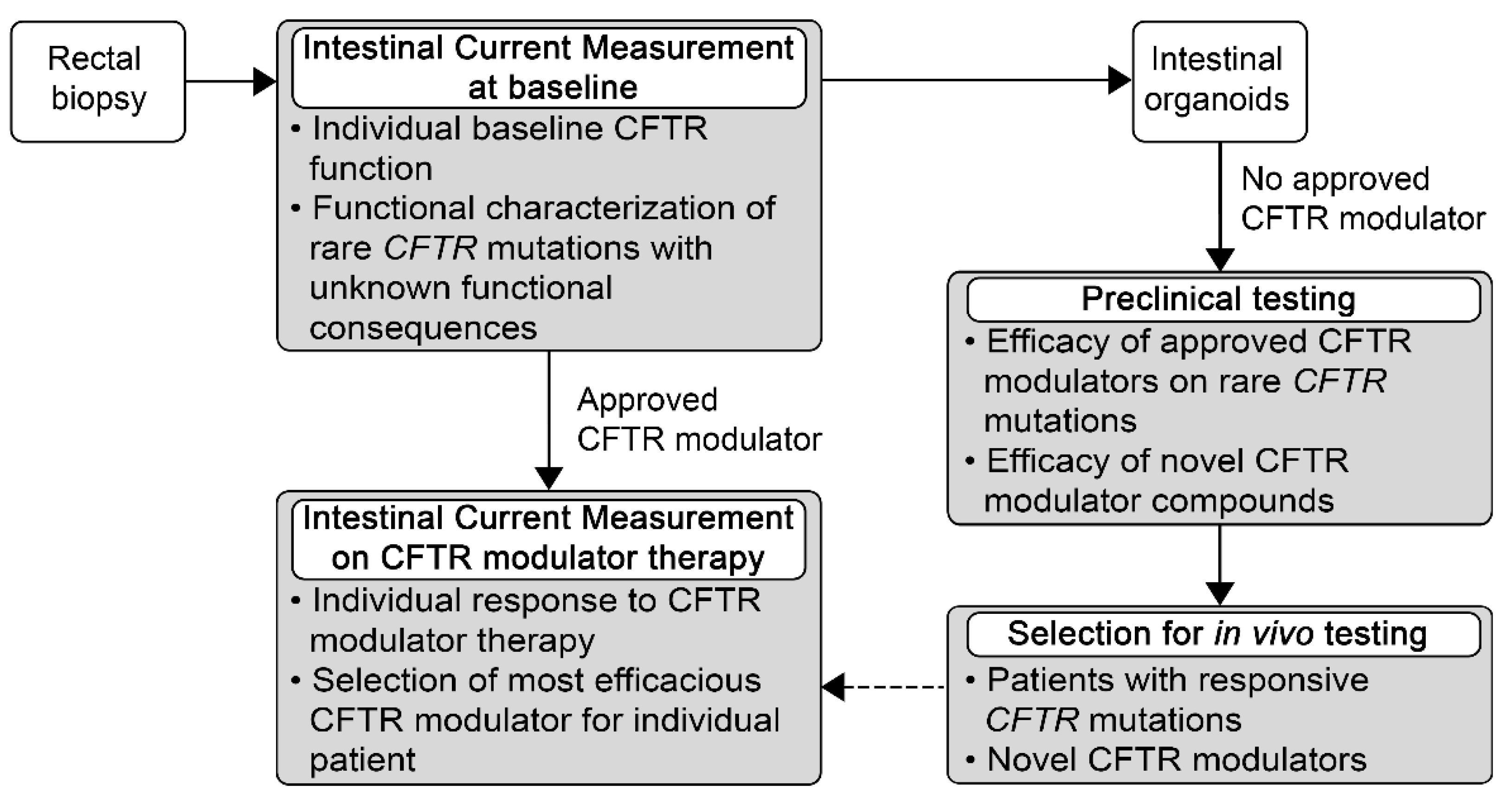
5. Potential Use of ICM for Personalized Medicine for Patients with Rare Mutations and High Unmet Need
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castanos, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Gentzsch, M.; Mall, M.A. Ion Channel Modulators in Cystic Fibrosis. Chest 2018, 154, 383–393. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; de Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.T.; Taylor-Cousar, J.L.; Davies, J.; Gibson, R.L.; Mall, M.A.; McKone, E.F.; McNally, P.; Ramsey, B.W.; Rayment, J.H.; Rowe, S.M.; et al. A Phase 3 Open-Label Study of ELX/TEZ/IVA in Children 6 through 11 Years of Age with CF and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Berschneider, H.M.; Knowles, M.R.; Azizkhan, R.G.; Boucher, R.C.; Tobey, N.A.; Orlando, R.C.; Powell, D.W. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988, 2, 2625–2629. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Baxter, P.S.; Hardcastle, J.; Hardcastle, P.T. Failure to induce secretion in jejunal biopsies from children with cystic fibrosis. Gut 1988, 29, 957–962. [Google Scholar] [CrossRef]
- Hardcastle, J.; Hardcastle, P.T.; Taylor, C.J.; Goldhill, J. Failure of cholinergic stimulation to induce a secretory response from the rectal mucosa in cystic fibrosis. Gut 1991, 32, 1035–1039. [Google Scholar] [CrossRef]
- Veeze, H.J.; Sinaasappel, M.; Bijman, J.; Bouquet, J.; de Jonge, H.R. Ion transport abnormalities in rectal suction biopsies from children with cystic fibrosis. Gastroenterology 1991, 101, 398–403. [Google Scholar] [CrossRef]
- Veeze, H.J.; Halley, D.J.; Bijman, J.; de Jongste, J.C.; de Jonge, H.R.; Sinaasappel, M. Determinants of mild clinical symptoms in cystic fibrosis patients. Residual chloride secretion measured in rectal biopsies in relation to the genotype. J. Clin. Investig. 1994, 93, 461–466. [Google Scholar] [CrossRef]
- Mall, M.; Bleich, M.; Greger, R.; Schreiber, R.; Kunzelmann, K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Investig. 1998, 102, 15–21. [Google Scholar] [CrossRef]
- Mall, M.; Bleich, M.; Schurlein, M.; Kuhr, J.; Seydewitz, H.H.; Brandis, M.; Greger, R.; Kunzelmann, K. Cholinergic ion secretion in human colon requires coactivation by cAMP. Am. J. Physiol. 1998, 275, G1274–G1281. [Google Scholar] [CrossRef]
- Mall, M.; Bleich, M.; Kuehr, J.; Brandis, M.; Greger, R.; Kunzelmann, K. CFTR-mediated inhibition of epithelial Na+ conductance in human colon is defective in cystic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 1999, 277, G709–G716. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Wissner, A.; Gonska, T.; Calenborn, D.; Kuehr, J.; Brandis, M.; Kunzelmann, K. Inhibition of amiloride-sensitive epithelial Na(+) absorption by extracellular nucleotides in human normal and cystic fibrosis airways. Am. J. Respir. Cell Mol. Biol. 2000, 23, 755–761. [Google Scholar] [CrossRef]
- Mall, M.; Wissner, A.; Seydewitz, H.H.; Kuehr, J.; Brandis, M.; Greger, R.; Kunzelmann, K. Defective cholinergic Cl(−) secretion and detection of K(+) secretion in rectal biopsies from cystic fibrosis patients. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G617–G624. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Wissner, A.; Seydewitz, H.H.; Hubner, M.; Kuehr, J.; Brandis, M.; Greger, R.; Kunzelmann, K. Effect of genistein on native epithelial tissue from normal individuals and CF patients and on ion channels expressed in Xenopus oocytes. Br. J. Pharmacol. 2000, 130, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Bronsveld, I.; Mekus, F.; Bijman, J.; Ballmann, M.; Greipel, J.; Hundrieser, J.; Halley, D.J.; Laabs, U.; Busche, R.; De Jonge, H.R.; et al. Residual chloride secretion in intestinal tissue of deltaF508 homozygous twins and siblings with cystic fibrosis. The European CF Twin and Sibling Study Consortium. Gastroenterology 2000, 119, 32–40. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mall, M. Electrolyte transport in the mammalian colon: Mechanisms and implications for disease. Physiol. Rev. 2002, 82, 245–289. [Google Scholar] [CrossRef]
- Hirtz, S.; Gonska, T.; Seydewitz, H.H.; Thomas, J.; Greiner, P.; Kuehr, J.; Brandis, M.; Eichler, I.; Rocha, H.; Lopes, A.I.; et al. CFTR Cl-channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 2004, 127, 1085–1095. [Google Scholar] [CrossRef]
- Mall, M.; Hirtz, S.; Gonska, T.; Kunzelmann, K. Assessment of CFTR function in rectal biopsies for the diagnosis of cystic fibrosis. J. Cyst. Fibros. 2004, 3 (Suppl. 2), 165–169. [Google Scholar] [CrossRef]
- Mall, M.; Kreda, S.M.; Mengos, A.; Jensen, T.J.; Hirtz, S.; Seydewitz, H.H.; Yankaskas, J.; Kunzelmann, K.; Riordan, J.R.; Boucher, R.C. The DeltaF508 mutation results in loss of CFTR function and mature protein in native human colon. Gastroenterology 2004, 126, 32–41. [Google Scholar] [CrossRef]
- De Jonge, H.R.; Ballmann, M.; Veeze, H.; Bronsveld, I.; Stanke, F.; Tummler, B.; Sinaasappel, M. Ex vivo CF diagnosis by intestinal current measurements (ICM) in small aperture, circulating Ussing chambers. J. Cyst. Fibros. 2004, 3 (Suppl. 2), 159–163. [Google Scholar] [CrossRef]
- Frizzell, R.A.; Rechkemmer, G.; Shoemaker, R.L. Altered regulation of airway epithelial cell chloride channels in cystic fibrosis. Science 1986, 233, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Clarke, L.L.; Boucher, R.C. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N. Engl. J. Med. 1991, 325, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Welsh, M.J. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proc. Natl. Acad. Sci. USA 1991, 88, 6003–6007. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.D.; Schreur, K.D.; Ji, H.L.; Fuller, C.M.; Pauli, B.U. Molecular cloning and transmembrane structure of hCLCA2 from human lung, trachea, and mammary gland. Am. J. Physiol. 1999, 276, C1261–C1270. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.; Boucher, R. Chloride secretory response to extracellular ATP in human normal and cystic fibrosis nasal epithelia. Am. J. Physiol. Cell Physiol. 1992, 263, C348–C356. [Google Scholar] [CrossRef]
- Mall, M.; Gonska, T.; Thomas, J.; Schreiber, R.; Seydewitz, H.H.; Kuehr, J.; Brandis, M.; Kunzelmann, K. Modulation of Ca2+-activated Cl− secretion by basolateral K+ channels in human normal and cystic fibrosis airway epithelia. Pediatr. Res. 2003, 53, 608–618. [Google Scholar] [CrossRef]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Salomon, J.J.; Albrecht, T.; Graeber, S.Y.; Scheuermann, H.; Butz, S.; Schatterny, J.; Mairbaurl, H.; Baumann, I.; Mall, M.A. Chronic rhinosinusitis with nasal polyps is associated with impaired TMEM16A-mediated epithelial chloride secretion. J. Allergy Clin. Immunol. 2021. [Google Scholar] [CrossRef]
- Derichs, N.; Sanz, J.; Von Kanel, T.; Stolpe, C.; Zapf, A.; Tummler, B.; Gallati, S.; Ballmann, M. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: Validation and reference data. Thorax 2010, 65, 594–599. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Kent, L.; Davies, J.; Derichs, N.; Amaral, M.; Rowe, S.M.; Middleton, P.; de Jonge, H.; Bronsveld, I.; Wilschanski, M.; et al. CFTR biomarkers: Time for promotion to surrogate end-point. Eur. Respir. J. 2013, 41, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Yaakov, Y.; Omari, I.; Zaman, M.; Martin, C.R.; Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E.; Dasilva, D.; Sheth, S.; et al. Comparison of Nasal Potential Difference and Intestinal Current Measurements as Surrogate Markers for CFTR Function. J. Pediatr. Gastroenterol. Nutr. 2016, 63, e92–e97. [Google Scholar] [CrossRef] [PubMed]
- Roth, E.K.; Hirtz, S.; Duerr, J.; Wenning, D.; Eichler, I.; Seydewitz, H.H.; Amaral, M.D.; Mall, M.A. The K+ channel opener 1-EBIO potentiates residual function of mutant CFTR in rectal biopsies from cystic fibrosis patients. PLoS ONE 2011, 6, e24445. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Hug, M.J.; Sommerburg, O.; Hirtz, S.; Hentschel, J.; Heinzmann, A.; Dopfer, C.; Schulz, A.; Mainz, J.G.; Tummler, B.; et al. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am. J. Respir. Crit. Care Med. 2015, 192, 1252–1255. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Dopfer, C.; Naehrlich, L.; Gyulumyan, L.; Scheuermann, H.; Hirtz, S.; Wege, S.; Mairbaurl, H.; Dorda, M.; Hyde, R.; et al. Effects of Lumacaftor-Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1433–1442. [Google Scholar] [CrossRef]
- Masson, A.; Schneider-Futschik, E.K.; Baatallah, N.; Nguyen-Khoa, T.; Girodon, E.; Hatton, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; et al. Predictive factors for lumacaftor/ivacaftor clinical response. J. Cyst. Fibros. 2019, 18, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.; Servidoni, M.F.; Vinagre, A.M.; Ramalho, A.S.; Bonadia, L.C.; Felicio, V.; Ribeiro, M.A.; Uliyakina, I.; Marson, F.A.; Kmit, A.; et al. Measurements of CFTR-mediated Cl− secretion in human rectal biopsies constitute a robust biomarker for Cystic Fibrosis diagnosis and prognosis. PLoS ONE 2012, 7, e47708. [Google Scholar] [CrossRef] [PubMed]
- Beekman, J.M.; Sermet-Gaudelus, I.; de Boeck, K.; Gonska, T.; Derichs, N.; Mall, M.A.; Mehta, A.; Martin, U.; Drumm, M.; Amaral, M.D. CFTR functional measurements in human models for diagnosis, prognosis and personalized therapy: Report on the pre-conference meeting to the 11th ECFS Basic Science Conference, Malta, 26–29 March 2014. J. Cyst. Fibros. 2014, 13, 363–372. [Google Scholar] [CrossRef][Green Version]
- Cohen-Cymberknoh, M.; Yaakov, Y.; Shoseyov, D.; Shteyer, E.; Schachar, E.; Rivlin, J.; Bentur, L.; Picard, E.; Aviram, M.; Israeli, E. Evaluation of the intestinal current measurement method as a diagnostic test for cystic fibrosis. Pediatr. Pulmonol. 2013, 48, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Szczesniak, R.D.; Ashlock, M.A.; Ernst, S.E.; Fan, L.; Hornick, D.B.; Karp, P.H.; Khan, U.; Lymp, J.; Ostmann, A.J.; et al. Multicenter intestinal current measurements in rectal biopsies from CF and non-CF subjects to monitor CFTR function. PLoS ONE 2013, 8, e73905. [Google Scholar] [CrossRef]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181S, S4–S15.e11. [Google Scholar] [CrossRef] [PubMed]
- Minso, R.; Schulz, A.; Dopfer, C.; Alfeis, N.; Barneveld, A.V.; Makartian-Gyulumyan, L.; Hansen, G.; Junge, S.; Muller, C.; Ringshausen, F.C.C.; et al. Intestinal current measurement and nasal potential difference to make a diagnosis of cases with inconclusive CFTR genetics and sweat test. BMJ Open Respir. Res. 2020, 7. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Duarte, A.; Marson, F.A.L.; Centeio, R.; Doušová, T.; Kunzelmann, K.; Amaral, M.D. Assessment of Distinct Electrophysiological Parameters in Rectal Biopsies for the Choice of the Best Diagnosis/Prognosis Biomarkers for Cystic Fibrosis. Front. Physiol. 2020, 11, 604580. [Google Scholar] [CrossRef]
- Sermet-Gaudelus, I.; Brouard, J.; Audrézet, M.P.; Couderc Kohen, L.; Weiss, L.; Wizla, N.; Vrielynck, S.; Llerena, K.; Le Bourgeois, M.; Deneuville, E.; et al. Guidelines for the clinical management and follow-up of infants with inconclusive cystic fibrosis diagnosis through newborn screening. Arch. Pediatr. 2017, 24, e1–e14. [Google Scholar] [CrossRef]
- Barben, J.; Castellani, C.; Munck, A.; Davies, J.C.; de Winter-de Groot, K.M.; Gartner, S.; Kashirskaya, N.; Linnane, B.; Mayell, S.J.; McColley, S.; et al. Updated guidance on the management of children with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome/cystic fibrosis screen positive, inconclusive diagnosis (CRMS/CFSPID). J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Lindner, M.; Muckenthaler, M.; Kohlmueller, D.; Leible, S.; Feneberg, R.; Kulozik, A.E.; Mall, M.A.; Hoffmann, G.F. Initial evaluation of a biochemical cystic fibrosis newborn screening by sequential analysis of immunoreactive trypsinogen and pancreatitis-associated protein (IRT/PAP) as a strategy that does not involve DNA testing in a Northern European population. J. Inherit. Metab. Dis. 2010, 33, S263–S271. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Krulisova, V.; Hammermann, J.; Lindner, M.; Stahl, M.; Muckenthaler, M.; Kohlmueller, D.; Happich, M.; Kulozik, A.E.; Votava, F.; et al. Comparison of different IRT-PAP protocols to screen newborns for cystic fibrosis in three central European populations. J. Cyst. Fibros. 2014, 13, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Awatade, N.T.; Uliyakina, I.; Farinha, C.M.; Clarke, L.A.; Mendes, K.; Solé, A.; Pastor, J.; Ramos, M.M.; Amaral, M.D. Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis. EBioMedicine 2015, 2, 147–153. [Google Scholar] [CrossRef]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef] [PubMed]
- Mutyam, V.; Libby, E.F.; Peng, N.; Hadjiliadis, D.; Bonk, M.; Solomon, G.M.; Rowe, S.M. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J. Cyst. Fibros. 2017, 16, 24–29. [Google Scholar] [CrossRef]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; De Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Moraes, T.J.; He, G.; Bartlett, C.; Szàrics, I.; Ouyang, H.; Gunawardena, T.N.A.; Strug, L.; Bear, C.E.; Gonska, T. The CFTR Mutation c.3453G > C (D1152H) Confers an Anion Selectivity Defect in Primary Airway Tissue That Can Be Rescued by Ivacaftor. J. Pers. Med. 2020, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; McCormack, J.; Bartlett, C.; Ip, W.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Gonska, T.; Moraes, T.J.; Bear, C.E. Preclinical Studies of a Rare CF-Causing Mutation in the Second Nucleotide Binding Domain (c.3700A>G) Show Robust Functional Rescue in Primary Nasal Cultures by Novel CFTR Modulators. J. Pers. Med. 2020, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Phuan, P.W.; Haggie, P.M.; Tan, J.A.; Rivera, A.A.; Finkbeiner, W.E.; Nielson, D.W.; Thomas, M.M.; Janahi, I.A.; Verkman, A.S. CFTR modulator therapy for cystic fibrosis caused by the rare c.3700A>G mutation. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Laselva, O.; Eckford, P.D.W.; Bartlett, C.; Ouyang, H.; Gunawardena, T.N.A.; Gonska, T.; Moraes, T.J.; Bear, C.E. Functional rescue of c.3846G>A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action. J. Cyst. Fibros. 2020, 19, 717–727. [Google Scholar] [CrossRef]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.S.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. Orkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor + tezacaftor + ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.A.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros. 2021, 20, 106–119. [Google Scholar] [CrossRef] [PubMed]
- de Winter-de Groot, K.M.; Janssens, H.M.; van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of Organoid Swelling and In Vivo Biomarkers of CFTR Function to Determine Effects of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- Berkers, G.; van der Meer, R.; Heijerman, H.; Beekman, J.M.; Boj, S.F.; Vries, R.G.J.; van Mourik, P.; Doyle, J.R.; Audhya, P.; Yuan, Z.J.; et al. Lumacaftor/ivacaftor in people with cystic fibrosis with an A455E-CFTR mutation. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef]
- Aalbers, B.L.; Hofland, R.W.; Bronsveld, I.; de Winter-de Groot, K.M.; Arets, H.G.M.; de Kiviet, A.C.; van Oirschot-van de Ven, M.M.M.; Kruijswijk, M.A.; Schotman, S.; Michel, S.; et al. Females with cystic fibrosis have a larger decrease in sweat chloride in response to lumacaftor/ivacaftor compared to males. J. Cyst. Fibros. 2021, 20, e7–e11. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Boutin, S.; Wielputz, M.O.; Joachim, C.; Frey, D.L.; Wege, S.; Sommerburg, O.; Kauczor, H.U.; Stahl, M.; Dalpke, A.H.; et al. Effects of Lumacaftor-Ivacaftor on Lung Clearance Index, Magnetic Resonance Imaging and Airway Microbiome in Phe508del Homozygous Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2021. [Google Scholar] [CrossRef]
- Bagheri-Hanson, A.; Nedwed, S.; Rueckes-Nilges, C.; Naehrlich, L. Intestinal current measurement versus nasal potential difference measurements for diagnosis of cystic fibrosis: A case-control study. BMC Pulm. Med. 2014, 14, 156. [Google Scholar] [CrossRef]


{kind=link}
| Category | Reference |
|---|---|
| Development of ICM | [15,16,17,18,19,21,22,24,26] |
| ICM as a diagnostic test for CF | [28,29,30,31,40,42,47,49,50,53,54,75,80] |
| ICM as outcome measure of preclinical ex vivo response to CFTR-directed therapeutics | [25,43,48] |
| ICM as outcome measure of in vivo response to CFTR-directed therapeutics | [44,45,46,76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graeber, S.Y.; Vitzthum, C.; Mall, M.A. Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis. J. Pers. Med. 2021, 11, 384. https://doi.org/10.3390/jpm11050384
Graeber SY, Vitzthum C, Mall MA. Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis. Journal of Personalized Medicine. 2021; 11(5):384. https://doi.org/10.3390/jpm11050384
Chicago/Turabian StyleGraeber, Simon Y., Constanze Vitzthum, and Marcus A. Mall. 2021. "Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis" Journal of Personalized Medicine 11, no. 5: 384. https://doi.org/10.3390/jpm11050384
APA StyleGraeber, S. Y., Vitzthum, C., & Mall, M. A. (2021). Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis. Journal of Personalized Medicine, 11(5), 384. https://doi.org/10.3390/jpm11050384