1. Introduction
After metformin, sulfonylureas (SUs) are the most commonly prescribed drugs in the treatment of type 2 diabetes (T2D) worldwide [
1]. This is due to their high efficacy and least cost, despite the development of newer antidiabetic agents. SUs are particularly widely used in low and middle-income countries, where they are recommended as the treatment of choice for T2D when metformin monotherapy fails to achieve glycemic targets [
2].
Hypoglycemia is the most common and potentially most serious side effect of sulfonylurea treatment [
3]. Severe hypoglycemia is associated with significantly increased morbidity and mortality [
4,
5,
6]. Among different SUs, gliclazide is recommended as the treatment of choice in many countries, as it seems to be associated with a lower risk of hypoglycemia and lower morbidity and mortality compared with other SUs [
7,
8]. Furthermore, gliclazide has been included in the WHO Model List of Essential Medicines, based on its safety data in elderly patients. However, in a large-population-based cohort study, gliclazide showed a similar risk of hypoglycemia compared with other SUs [
9].
Gliclazide is extensively metabolized in the liver to inactive metabolites. Although CYP2C9 enzyme is involved in its metabolism [
10], gliclazide pharmacokinetics (PK) seem to be affected by
CYP2C19 genetic polymorphisms [
11,
12]. Observed gliclazide AUC was 3.4-fold [
11] and 5.1-fold [
12] higher in CYP2C19 poor metabolizers (PM) compared to normal metabolizers (NM) following single oral administration of gliclazide in healthy Chinese volunteers.
Gastrointestinal (GI) problems, such as gastroesophageal reflux disease, are common in patients with T2D [
13]. The prevalence of GI symptoms in those patients could be as high as 40% [
13]. Considering that about 70% of patients with diabetes use oral hypoglycemic agents, millions of individuals are exposed to concomitant use of oral antidiabetic and acid-suppressing medications [
14]. Proton pump inhibitors (PPIs) are the treatment of choice for gastroesophageal reflux disease and are primarily metabolized by CYP2C19 [
15]. The potential for drug-drug interactions (DDI) is highest for omeprazole and its stereo-isomer esomeprazole, both metabolized almost entirely by CYP2C19 [
15,
16]. Furthermore, the impact of PPIs on the PK, pharmacodynamics, and therapeutic response of drugs metabolized by CYP2C19 was shown to depend on
CYP2C19 genotype, with the highest effect seen in CYP2C19 NM [
17,
18] and ultrarapid metabolizers (UM) [
19].
Physiologically based pharmacokinetic (PBPK) modeling is well established in the pharmaceutical industry and is accepted by regulatory agencies for the prediction of DDIs of new drugs [
20,
21,
22]. It has been widely used especially for CYP-mediated DDIs [
23]. Considering the possibility of interaction via CYP2C19, we aimed to explore the impact of omeprazole on gliclazide PK, in relation to CYP2C19 phenotype using a PBPK approach. Developed PBPK models were validated using in vivo PK profiles obtained in a clinical trial on omeprazole-gliclazide interaction in healthy volunteers, CYP2C19 normal/rapid/ultrarapid metabolizers (NM/RM/UM). Finally, the possible interaction between the two drugs was explored in the cohort of patients with T2D treated with gliclazide.
4. Discussion
This is the first study that explored the possible interaction between omeprazole and gliclazide. We used PBPK modeling approach to predict the effects of omeprazole on gliclazide PK and validated developed models using data from the clinical trial in healthy volunteers. In addition, we explored the influence of omeprazole on risk of severe hypoglycemia in real-world patients with T2D treated with gliclazide. Our results collectively suggest that omeprazole can increase exposure to gliclazide and thus increase the risk of side effects in individuals treated concomitantly with these two drugs if CYP2C19 enzyme is functional.
We first developed a PBPK model for omeprazole. Although Simcyp has a built-in model for omeprazole and few PBPK models for omeprazole were published earlier [
27,
28,
29,
42], the models did not perform well in the current version of Simcyp. Among others, one of the reasons is different and updated liver abundances of the enzymes. Considering that we were exploring interaction via polymorphic CYP2C19, we updated the phenotype frequencies of CYP2C9 and CYP2C19 enzymes and changed their abundancies in the liver according to the estimated enzyme activity scores for each phenotype. As the intrinsic clearance (Cl
int) values for enzymes depend on their abundancies, they were back calculated based on in vivo clearance data. Although it is better to use in vivo clearance from i.v. administration in the retrograde model for Cl
int calculations, we used oral clearance value as it was reported for subjects with known CYP2C19 phenotype (NM/RM/UM phenotype), and gut metabolism of omeprazole is expected to be negligible [
43]. The non-linear PK of omeprazole was previously best captured by published models for omeprazole enantiomers [
28]. We managed to optimise values for parameters related to mechanism-based inhibition of CYP2C19 by omeprazole (K
app and k
inact) using published data for enantiomers as starting values [
28]. The simulation of changes in the amount of active CYP2C19 following 5-day dosing with 40 mg omeprazole once daily showed that the lowest level of active CYP2C19 was 11% of baseline, in line with the predicted lowest level of 14% obtained in a previous study based on enantiomers [
28]. The model simulated well four drug plasma concentration-time profiles, as well as C
max and AUC values following single and multiple oral administration of 20 mg and 40 mg omeprazole. All predicted PK parameters were within 1.1-fold of the clinical data.
The model for gliclazide was developed using in vitro, in silico and in vivo data as inputs. As gliclazide is a substrate of liver OATP1B1 transporter [
37], which can contribute to its liver uptake and hence the metabolism and elimination, the Cl
int values for enzyme kinetics could not be back calculated from the in vivo data. Only one in vitro study of gliclazide metabolism was published using human liver microsomes and a panel of recombinant human CYPs [
10]. Based on the total microsomal intrinsic clearance, and the percentage of inhibition of metabolic pathways by CYP selective inhibitors, the contribution of CYP2C9 to gliclazide metabolism was estimated to be at least 65%, and CYP2C19 maximally 35% according to this study [
10]. However, microsomes were prepared from six human livers which were not genotyped for CYP2C19 polymorphisms. Furthermore, later in vivo studies showed that gliclazide PK is affected mainly by CYP2C19 polymorphisms [
11,
12,
55]. Based on these PK clinical studies in individuals with different CYP2C19 and CYP2C9 genotypes [
11,
12], it was estimated that the contribution of CYP2C9 to gliclazide metabolism is around 24%, and of CYP2C19 around 76% [
36]. Taking this into account, the Cl
int values for enzymes were calculated based on the overall in vitro microsomal intrinsic clearance [
10], and f
m values estimated from in vivo studies [
36].
By using enzyme Cl
int values only, clearance of the drug after i.v. administration was largely underpredicted. Therefore, we extended our model to include in vitro obtained values for OATP1B1-mediated gliclazide transport [
37], and optimized RAF value for the transporter to fit the observed data after i.v. drug administration [
35], as well as after oral drug dosing [
35,
44]. Namely, only one study describing gliclazide PK after i.v. administration was found in the literature and referred to i.v. infusion [
35], so oral drug dosing studies were also taken into account. The RAF or relative expression factor (REF) frequently needs to be optimized in the PBPK models due to a difference in expression of the transporter between liver and the transfected cell line. The RAF value of 5.75 was in line with bottom-up derived REF values of 5.9 and 8.7 for different OATP1B1 substrates [
56] based on measured OATP1B1 expression in the liver [
57], isolated hepatocytes [
57], and transfected HEK-293 cells in other studies [
56].
Another parameter that had to be optimized was MRP2-mediated intrinsic clearance as t
max values for oral formulations were underpredicted in the preliminary simulations. It has been shown that gliclazide is a substrate for gut Mrp2 and Mrp3 transporters in rats [
41], but in vitro and human data are lacking. As intestinal basolateral transporters are not yet included in the Simcyp ADAM model, only apical transporter MRP2 was added to the model. After this adjustment, the simulated t
max values were in accordance with the observed data after oral drug dosing.
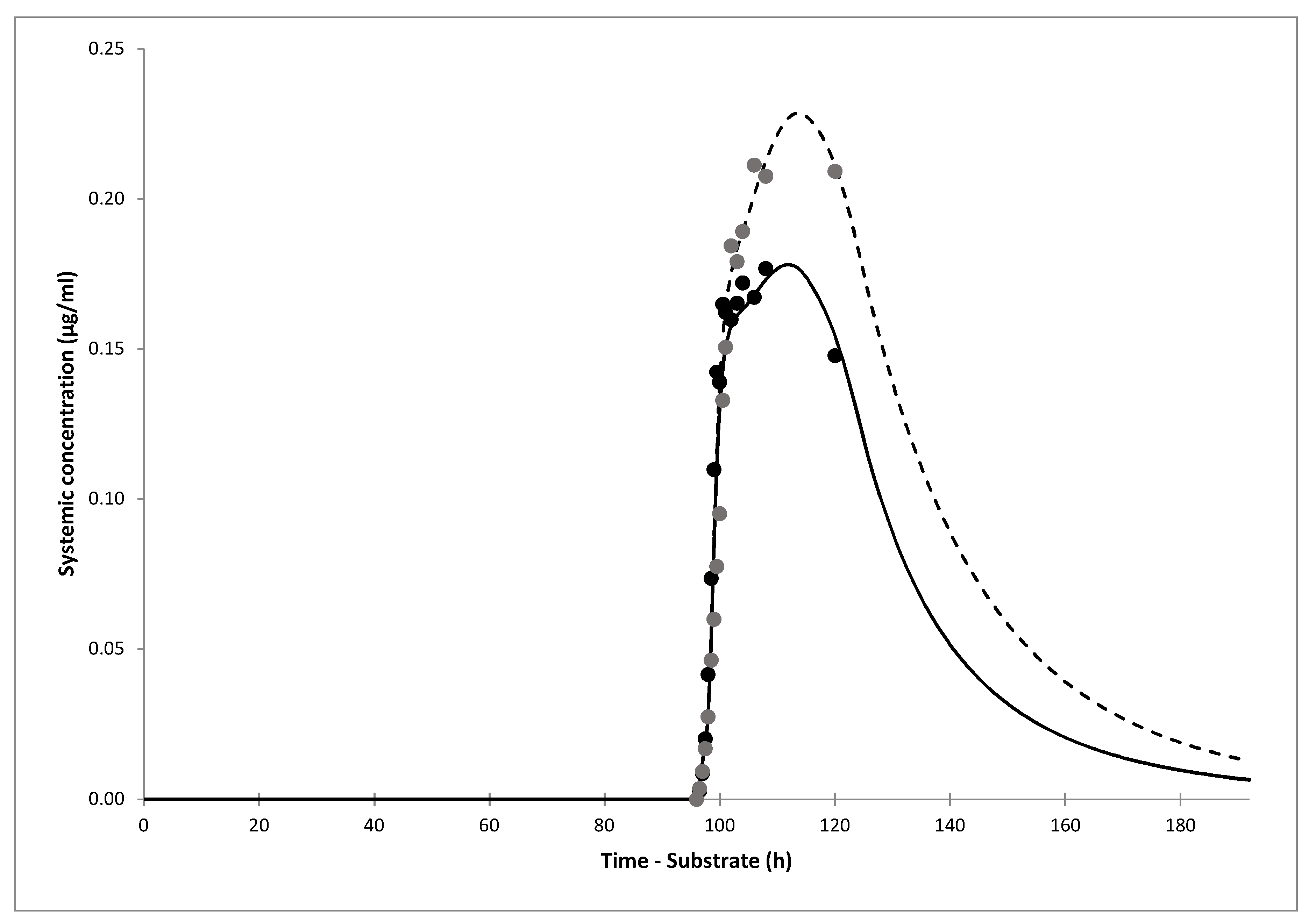
The simulations of gliclazide PK showed good agreement with clinical data after i.v. infusion, and oral administration of gliclazide IR and MR tablets. The model showed that only 30% (19–43%) of gliclazide was absorbed in our current clinical trial. The simulation of the interaction showed that the predicted DDI AUC ratio of gliclazide between omeprazole and placebo phases was 1.40 (1.35–1.45). The predicted ratio would have been 1.50 (1.44–1.56) if IR tablets with complete absorption were administered. The predicted DDI Cmax ratio for the current trial was 1.26 (1.23–1.29), however, Cmax would not have been impacted significantly by omeprazole dosing if gliclazide IR tablets were administered, according to the model.
The simulated DDI AUC ratio after 5-day dosing with 20 mg omeprazole was 1.55, 1.49, 1.40, and 1.01 for NM, RM/UM, IM and PM individuals, respectively. A slightly lower AUC ratio in RM/UM compared with NM individuals was due to lower predicted omeprazole systemic and liver concentrations and thus weaker inhibitory effect on CYP2C19. The lowest simulated percentage of active CYP2C19 was 31% in RM/UM compared to 21% in NM individuals. On the other hand, AUC ratio of 1.40 in IM individuals was comparable to the ratios obtained in NM and RM/UM phenotypes. Although simulated CYP2C19 fm value for gliclazide in IM individuals was only 59%, the interaction effect was similar to the one in the NM/RM/UM group, due to higher predicted systemic concentrations of omeprazole. As expected, no interaction effect in PM subjects was found due to low or absent CYP2C19.
Although the predicted omeprazole-gliclazide interaction effect was not large, when we simulated clinical scenarios of concomitant multiple dosing of both drugs for 20 days in NM/RM/UM individuals, the DDI AUC ratio increased to 2.57 (2.39–2.76) if 40 mg omeprazole was administered once daily, and to 3.80 (3.48–4.15) if it was administered twice daily. The predicted DDI AUC ratio in IM individuals also increased to 2.13 (2.00–2.27) and 2.53 (2.35–2.73) after co-administration of 40 mg omeprazole once and twice daily, respectively.
We used the Simcyp population of healthy volunteers for all simulations, as there is no special built-in population of patients with T2D in the Simcyp. However, considering the older age of these patients and possible age-related PK changes such as limited drug clearance due to decreased liver blood flow and reduced kidney function, as well as frequent concomitant therapy with other interacting drugs, the concentrations of drugs and thus interaction effect can be greater in older patients compared to young healthy volunteers [
58]. Interestingly, a recent study found lower mean metabolic activities of CYP2C19, CYP2B6, and CYP3A enzymes in patients with T2D compared to non-T2D individuals [
59]. However, although lower activity of CYP2C19 can decrease the CYP2C19 f
m value for gliclazide, the inhibition of CYP21C9 could be stronger due to higher systemic concentrations of omeprazole. In line with this, in a study that explored the effect of ageing on omeprazole PK, the omeprazole AUC was higher in elderly compared to young Japanese volunteers following single i.v. dose of omeprazole [
60].
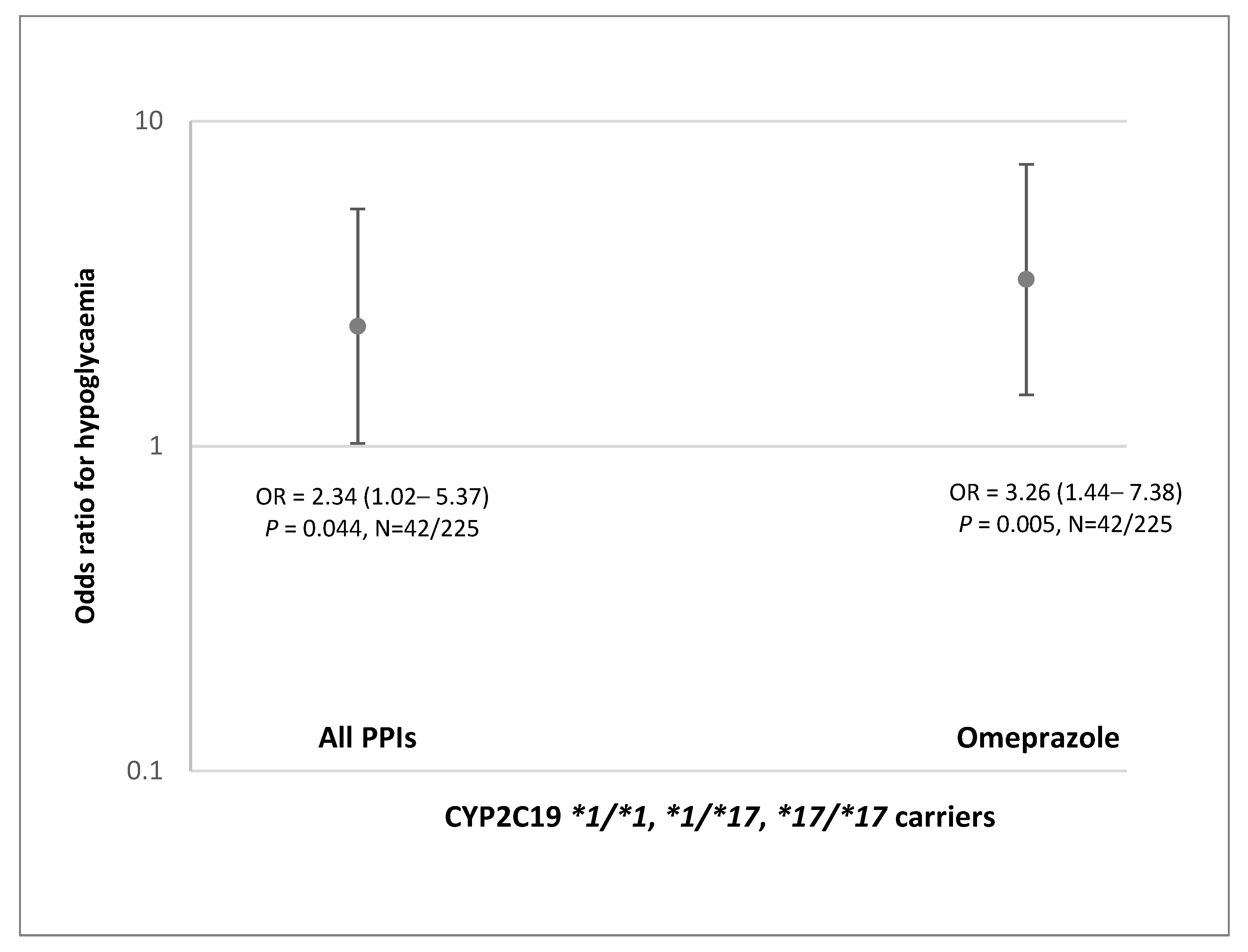
Finally, we explored the effect of co-treatment with omeprazole and other PPI drugs in real-world patients with T2D treated with gliclazide. Although patient cohort with available genetic data was not large, we showed that patients with CYP2C19 NM/RM/UM phenotype treated with omeprazole had 3.26-fold (1.44–7.38) higher odds of severe hypoglycemia. These results corroborate higher systemic exposure to gliclazide predicted for NM/RM/UM individuals. No effect was seen in a very small group of patients carrying CYP2C19*2 allele.
To our best knowledge, there have been no studies exploring potential interaction between any PPI and gliclazide. Data regarding potential DDI between PPIs and other SUs in humans are also limited. One study conducted in healthy volunteers showed no effect of pantoprazole on glibenclamide PK [
61]. As glibenclamide is metabolized by CYP2C9 and CYP3A4, and other SUs, including glimepiride, glipizide and gliquidone, are metabolized mostly by CYP2C9 [
62], potential PK interactions with PPIs are not expected. On the other hand, other PPIs could also interact with gliclazide via CYP2C19, however, except for esomeprazole, their potential for DDIs seems to be weaker [
15].
The main limitation of our study are incomplete in vivo gliclazide PK profiles in omeprazole and placebo phase obtained in the current clinical trial. After we informed the Agency for Medicines and Medical Devices of Bosnia and Herzegovina about the unexpected MR profile of Diprian® tablets, the drug was withdrawn from the market and thus patients in B&H stopped receiving this possibly inefficient drug. Still, we had collected data up to 24 h, when drug elimination had mostly prevailed, and PK profiles in both phases were well simulated. Our PBPK models confirmed the mechanistic hypothesis of the interaction between the two drugs via polymorphic CYP2C19 enzyme and also allowed us to predict different clinical scenarios.
Our results have potential clinical relevance. The predicted increase in gliclazide AUC after 20-day of concomitant dosing with omeprazole shows that NM/RM/UM individuals may have almost 4-times higher exposure to gliclazide. This is in line with over 3-fold higher odds of hypoglycemia seen in NM/RM/UM patients with T2D treated concomitantly with omeprazole and gliclazide. The predicted AUC increase in IM individuals was lower but still over 2-fold. This may lead to an increased risk of gliclazide side effects in all patients treated with omeprazole, except those with the CYP2C19 PM phenotype. The risk may be even higher if other clinical risk factors for hypoglycemia are present. Avoiding co-prescribing omeprazole in these patients may prevent serious consequences of severe hypoglycemia induced by gliclazide treatment.
In conclusion, we applied PBPK models to simulate the clinical PK profiles and to assess the potential interaction between omeprazole and gliclazide. Developed models predicted 1.4–1.6-fold higher gliclazide AUC in all CYP2C19 phenotypes except PM, after 5-day treatment with 20 mg omeprazole. The predicted gliclazide AUC increased 2.1 and 2.5-fold in IM individuals, and 2.6 and 3.8-fold in the NM/RM/UM group after 20 days of concomitant gliclazide administration with 40 mg omeprazole once and twice daily, respectively. The predicted results were corroborated by findings in patients with T2D which demonstrated over 3-fold higher odds of severe gliclazide-induced hypoglycemia in patients with CYP2C19 NM/RM/UM phenotype treated with omeprazole. These results indicate that omeprazole may increase the risk of hypoglycemia in the majority of patients treated with gliclazide. Further studies are needed to confirm this previously unrecognized but potentially serious DDI and drug–drug–gene interaction.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




