
Genetic Predictors for Sinusoidal Obstruction Syndrome—A Systematic Review

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Study Selection: Eligibility Criteria
2.3. Outcome Definition: Sinusoidal Obstruction Syndrome
2.4. Exposures: Genetic Variants
2.5. Identification of Studies
2.6. Study Selection
2.7. Data Extraction
2.8. Quality Assessment and Risk of Bias
3. Results
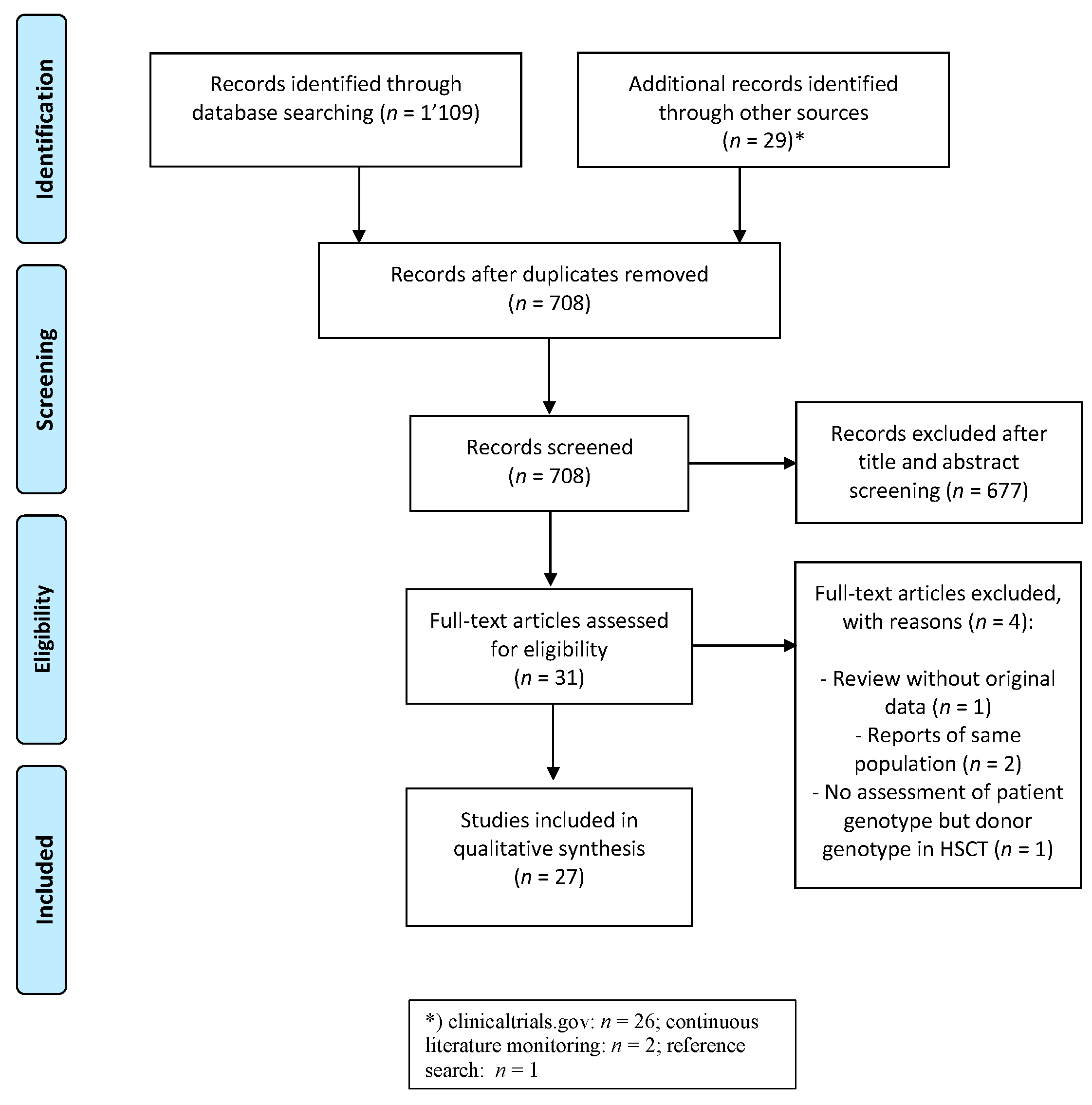
3.1. Study Identification and Selection
3.2. Characteristics of Included Studies
3.2.1. Study Characteristics
3.2.2. Population
3.2.3. Treatment Exposure
3.2.4. Genotyping
3.2.5. Outcome
3.3. Quality of Studies and Publication Bias
3.4. Investigated Genes for Association with SOS after HSCT
3.4.1. Glutathione S-Transferase
3.4.2. Cytochrome P450
3.4.3. Methylenetetrahydrofolate Reductase
3.4.4. Other Liver Enzymes
3.4.5. Coagulation and Vascular System
3.4.6. Whole Exome Analysis
3.5. Investigated Genes for Association with Antineoplastic Agent Exposure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahadeo, K.M.; Bajwa, R.; Abdel-Azim, H.; Lehmann, L.E.; Duncan, C.; Zantek, N.; Vittorio, J.; Angelo, J.; McArthur, J.; Schadler, K.; et al. Diagnosis, Grading, and Treatment Recommendations for Children, Adolescents, and Young Adults with Sinusoidal Obstructive Syndrome: An International Expert Position Statement. Lancet Haematol. 2020, 7, e61–e72. [Google Scholar] [CrossRef]
- Fan, C.Q.; Crawford, J.M. Sinusoidal Obstruction Syndrome (Hepatic Veno-Occlusive Disease). J. Clin. Exp. Hepatol. 2014, 4, 332–346. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.B.; Sharma, P.; Matthews, D.E.; Shulman, H.M.; Thomas, E.D. Venocclusive Disease of the Liver after Bone Marrow Transplantation: Diagnosis, Incidence, and Predisposing Factors. Hepatology 1984, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Lee, K.S.; Beschorner, W.E.; Vogel, V.G.; Grochow, L.B.; Braine, H.G.; Vogelsang, G.B.; Sensenbrenner, L.L.; Santos, G.W.; Saral, R. Venoocclusive Disease of the Liver Following Bone Marrow Transplantation. Transplantation 1987, 44, 778–783. [Google Scholar] [CrossRef]
- Corbacioglu, S.; Carreras, E.; Ansari, M.; Balduzzi, A.; Cesaro, S.; Dalle, J.-H.; Dignan, F.; Gibson, B.; Guengoer, T.; Gruhn, B.; et al. Diagnosis and Severity Criteria for Sinusoidal Obstruction Syndrome/Veno-Occlusive Disease in Pediatric Patients: A New Classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2018, 53, 138–145. [Google Scholar] [CrossRef]
- Coppell, J.A.; Richardson, P.G.; Soiffer, R.; Martin, P.L.; Kernan, N.A.; Chen, A.; Guinan, E.; Vogelsang, G.; Krishnan, A.; Giralt, S.; et al. Hepatic Veno-Occlusive Disease Following Stem Cell Transplantation: Incidence, Clinical Course, and Outcome. Biol. Blood Marrow Transplant. 2010, 16, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Dalle, J.-H.; Giralt, S.A. Hepatic Veno-Occlusive Disease after Hematopoietic Stem Cell Transplantation: Risk Factors and Stratification, Prophylaxis, and Treatment. Biol. Blood Marrow Transplant. 2016, 22, 400–409. [Google Scholar] [CrossRef]
- McDonald, G.B.; Hinds, M.S.; Fisher, L.D.; Schoch, H.G.; Wolford, J.L.; Banaji, M.; Hardin, B.J.; Shulman, H.M.; Clift, R.A. Veno-Occlusive Disease of the Liver and Multiorgan Failure after Bone Marrow Transplantation: A Cohort Study of 355 Patients. Ann. Intern. Med. 1993, 118, 255–267. [Google Scholar] [CrossRef]
- Corbacioglu, S.; Cesaro, S.; Faraci, M.; Valteau-Couanet, D.; Gruhn, B.; Rovelli, A.; Boelens, J.J.; Hewitt, A.; Schrum, J.; Schulz, A.S.; et al. Defibrotide for Prophylaxis of Hepatic Veno-Occlusive Disease in Paediatric Haemopoietic Stem-Cell Transplantation: An Open-Label, Phase 3, Randomised Controlled Trial. Lancet 2012, 379, 1301–1309. [Google Scholar] [CrossRef]
- Choi, J.-H.; Won, Y.-W.; Kim, H.S.; Oh, Y.-H.; Lim, S.; Kim, H.-J. Oxaliplatin-Induced Sinusoidal Obstruction Syndrome Mimicking Metastatic Colon Cancer in the Liver. Oncol. Lett. 2016, 11, 2861–2864. [Google Scholar] [CrossRef]
- Koo, S.X.; Chan, S.H.; Ngeow, J. Genetic Predisposition Resulting in Sinusoidal Obstruction Syndrome in a Patient with Resected Sigmoid Cancer on Adjuvant Oxaliplatin. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef]
- Lennard, L.; Richards, S.; Cartwright, C.S.; Mitchell, C.; Lilleyman, J.S.; Vora, A. UK MRC/NCRI Childhood Leukaemia Working Party The Thiopurine Methyltransferase Genetic Polymorphism Is Associated with Thioguanine-Related Veno-Occlusive Disease of the Liver in Children with Acute Lymphoblastic Leukemia. Clin. Pharm. 2006, 80, 375–383. [Google Scholar] [CrossRef]
- Wray, L.; Vujkovic, M.; McWilliams, T.; Cannon, S.; Devidas, M.; Stork, L.; Aplenc, R. TPMT and MTHFR Genotype Is Not Associated with Altered Risk of Thioguanine-Related Sinusoidal Obstruction Syndrome in Pediatric Acute Lymphoblastic Leukemia: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 2086–2088. [Google Scholar] [CrossRef] [PubMed]
- Valla, D.-C.; Cazals-Hatem, D. Sinusoidal Obstruction Syndrome. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 378–385. [Google Scholar] [CrossRef]
- Kizilocak, H.; Dikme, G.; Özdemir, N.; Kuruğoğlu, S.; Adaletli, İ.; Erkan, T.; Celkan, T. Sinusoidal Obstruction Syndrome During Chemotherapy of Pediatric Cancers and Its Successful Management With Defibrotide. J. Pediatr. Hematol. Oncol. 2017, 39, e373–e376. [Google Scholar] [CrossRef] [PubMed]
- Mohty, M.; Malard, F.; Abecassis, M.; Aerts, E.; Alaskar, A.S.; Aljurf, M.; Arat, M.; Bader, P.; Baron, F.; Bazarbachi, A.; et al. Sinusoidal Obstruction Syndrome/Veno-Occlusive Disease: Current Situation and Perspectives—A Position Statement from the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. 2015, 50, 781–789. [Google Scholar] [CrossRef]
- Qiao, J.; Fu, J.; Fang, T.; Huang, Y.; Mi, H.; Yang, N.; Chen, C.; Xu, K.; Zeng, L. Evaluation of the Effects of Preconditioning Regimens on Hepatic Veno-Occlusive Disease in Mice after Hematopoietic Stem Cell Transplantation. Exp. Mol. Pathol. 2015, 98, 73–78. [Google Scholar] [CrossRef]
- Park, Y.D.; Yasui, M.; Yoshimoto, T.; Chayama, K.; Shimono, T.; Okamura, T.; Inoue, M.; Yumura-Yagi, K.; Kawa-Ha, K. Changes in Hemostatic Parameters in Hepatic Veno-Occlusive Disease Following Bone Marrow Transplantation. Bone Marrow Transplant. 1997, 19, 915–920. [Google Scholar] [CrossRef][Green Version]
- Carreras, E. Veno-Occlusive Disease of the Liver after Hemopoietic Cell Transplantation. Eur. J. Haematol. 2000, 64, 281–291. [Google Scholar] [CrossRef]
- Richardson, P.; Aggarwal, S.; Topaloglu, O.; Villa, K.F.; Corbacioglu, S. Systematic Review of Defibrotide Studies in the Treatment of Veno-Occlusive Disease/Sinusoidal Obstruction Syndrome (VOD/SOS). Bone Marrow Transplant. 2019, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Sartori, M.T.; Bisogno, G.; Van Schilfgaarde, M.; Saggiorato, G.; Pierro, A.M.D.; Corvetta, D.; Marcheselli, L.; Andrea, M.; Gastl, G.; et al. New Insights into Sinusoidal Obstruction Syndrome. Intern. Med. J. 2017, 47, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Duggan, C.; Schmidt, M.; Lawler, M.; White, B.; Cusack, S.; McCann, S.; Smith, O. The Prothrombin Gene Variant G20210A but Not Factor V Leiden May Be Associated with Veno-Occlusive Disease Following BMT. Bone Marrow Transplant. 1999, 24, 693–694. [Google Scholar] [CrossRef]
- Kim, M.G.; Kwak, A.; Choi, B.; Ji, E.; Oh, J.M.; Kim, K. Effect of Glutathione S-Transferase Genetic Polymorphisms on Busulfan Pharmacokinetics and Veno-Occlusive Disease in Hematopoietic Stem Cell Transplantation: A Meta-Analysis. Basic Clin. Pharmacol. Toxicol. 2019, 124, 691–703. [Google Scholar] [CrossRef]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols (PRISMA-P) 2015 Statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A Web and Mobile App for Systematic Reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef] [PubMed]
- Zazuli, Z.; Vijverberg, S.; Slob, E.; Liu, G.; Carleton, B.; Veltman, J.; Baas, P.; Masereeuw, R.; Maitland-van der Zee, A.-H. Genetic Variations and Cisplatin Nephrotoxicity: A Systematic Review. Front. Pharm. 2018, 9, 1111. [Google Scholar] [CrossRef] [PubMed]
- Little, J.; Higgins, J.P.T.; Ioannidis, J.P.A.; Moher, D.; Gagnon, F.; von Elm, E.; Khoury, M.J.; Cohen, B.; Davey-Smith, G.; Grimshaw, J.; et al. Strengthening the Reporting of Genetic Association Studies (STREGA): An Extension of the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement. J. Clin. Epidemiol. 2009, 62, 597–608.e4. [Google Scholar] [CrossRef]
- Vandenbroucke, J.; von Elm, E.; Altman, D.; Gøtzsche, P.; Mulrow, C.; Pocock, S.; Poole, C.; Schlesselman, J.; Egger, M. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE). Epidemiology 2007, 18, 805–835. [Google Scholar] [CrossRef]
- Ansari, M.; Lauzon-Joset, J.-F.; Vachon, M.-F.; Duval, M.; Théoret, Y.; Champagne, M.A.; Krajinovic, M. Influence of GST Gene Polymorphisms on Busulfan Pharmacokinetics in Children. Bone Marrow Transplant. 2010, 45, 261–267. [Google Scholar] [CrossRef]
- Ansari, M.; Rezgui, M.A.; Théoret, Y.; Uppugunduri, C.R.S.; Mezziani, S.; Vachon, M.-F.; Desjean, C.; Rousseau, J.; Labuda, M.; Przybyla, C.; et al. Glutathione S-Transferase Gene Variations Influence BU Pharmacokinetics and Outcome of Hematopoietic SCT in Pediatric Patients. Bone Marrow Transplant. 2013, 48, 939–946. [Google Scholar] [CrossRef]
- Piao, Z.; Kim, H.J.; Choi, J.Y.; Hong, C.R.; Lee, J.W.; Kang, H.J.; Park, K.D.; Shin, H.Y. Effect of FOXP3 Polymorphism on the Clinical Outcomes after Allogeneic Hematopoietic Stem Cell Transplantation in Pediatric Acute Leukemia Patients. Int. Immunopharmacol. 2016, 31, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Aplenc, R.; Vachani, A.; Han, P.; Glatfelter, W.; Sievers, E.L. Pharmacogenetics of Gemtuzumab-Associated Hepatic Sinusoidal Occlusion Syndrome after Hematopoietic Stem Cell Transplant. Acta Haematol. 2003, 110, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Efrati, E.; Zuckerman, T.; Ben-Ami, E.; Krivoy, N. MTHFR C677T/A1298C Genotype: A Possible Risk Factor for Liver Sinusoidal Obstruction Syndrome. Bone Marrow Transplant. 2014, 49, 726–727. [Google Scholar] [CrossRef] [PubMed]
- Elmaagacli, A.H.; Koldehoff, M.; Steckel, N.K.; Trenschel, R.; Ottinger, H.; Beelen, D.W. Cytochrome P 450 2C19 Loss-of-Function Polymorphism Is Associated with an Increased Treatment-Related Mortality in Patients Undergoing Allogeneic Transplantation. Bone Marrow Transplant. 2007, 40, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Kallianpur, A.R.; Hall, L.D.; Yadav, M.; Byrne, D.W.; Speroff, T.; Dittus, R.S.; Haines, J.L.; Christman, B.W.; Summar, M.L. The Hemochromatosis C282Y Allele: A Risk Factor for Hepatic Veno-Occlusive Disease after Hematopoietic Stem Cell Transplantation. Bone Marrow Transplant. 2005, 35, 1155–1164. [Google Scholar] [CrossRef]
- Terakura, S.; Onizuka, M.; Fukumoto, M.; Kuwatsuka, Y.; Kohno, A.; Ozawa, Y.; Miyamura, K.; Inagaki, Y.; Sawa, M.; Atsuta, Y.; et al. Analysis of Glutathione S-Transferase and Cytochrome P450 Gene Polymorphism in Recipients of Dose-Adjusted Busulfan-Cyclophosphamide Conditioning. Int. J. Hematol. 2020, 111, 84–92. [Google Scholar] [CrossRef]
- Krivoy, N.; Zuckerman, T.; Elkin, H.; Froymovich, L.; Rowe, J.M.; Efrati, E. Pharmacokinetic and Pharmacogenetic Analysis of Oral Busulfan in Stem Cell Transplantation: Prediction of Poor Drug Metabolism to Prevent Drug Toxicity. Curr. Drug Saf. 2012, 7, 211–217. [Google Scholar] [CrossRef]
- Kim, I.; Lee, K.-H.; Kim, J.H.; Ra, E.K.; Yoon, S.-S.; Hong, Y.-C.; Park, S.S.; Kim, C.S.; Park, S.; Kim, B.K. Polymorphisms of the Methylenetetrahydrofolate Reductase Gene and Clinical Outcomes in HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation. Ann. Hematol. 2007, 86, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Park, S.S.; Kim, I.; Kim, J.H.; Ra, E.K.; Yoon, S.-S.; Hong, Y.-C.; Park, S.; Kim, B.K. P2X7 Receptor Polymorphism and Clinical Outcomes in HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation. Haematologica 2007, 92, 651–657. [Google Scholar] [CrossRef]
- Rocha, V.; Porcher, R.; Fernandes, J.F.; Filion, A.; Bittencourt, H.; Silva, W.; Vilela, G.; Zanette, D.L.; Ferry, C.; Larghero, J.; et al. Association of Drug Metabolism Gene Polymorphisms with Toxicities, Graft-versus-Host Disease and Survival after HLA-Identical Sibling Hematopoietic Stem Cell Transplantation for Patients with Leukemia. Leukemia 2009, 23, 545–556. [Google Scholar] [CrossRef]
- Ansari, M.; Huezo-Diaz, P.; Rezgui, M.A.; Marktel, S.; Duval, M.; Bittencourt, H.; Cappelli, B.; Krajinovic, M. Influence of Glutathione S-Transferase Gene Polymorphisms on Busulfan Pharmacokinetics and Outcome of Hematopoietic Stem-Cell Transplantation in Thalassemia Pediatric Patients. Bone Marrow Transplant. 2016, 51, 377–383. [Google Scholar] [CrossRef]
- Srivastava, A.; Poonkuzhali, B.; Shaji, R.V.; George, B.; Mathews, V.; Chandy, M.; Krishnamoorthy, R. Glutathione S-Transferase M1 Polymorphism: A Risk Factor for Hepatic Venoocclusive Disease in Bone Marrow Transplantation. Blood 2004, 104, 1574–1577. [Google Scholar] [CrossRef]
- Uppugunduri, C.R.S.; Rezgui, M.A.; Diaz, P.H.; Tyagi, A.K.; Rousseau, J.; Daali, Y.; Duval, M.; Bittencourt, H.; Krajinovic, M.; Ansari, M. The Association of Cytochrome P450 Genetic Polymorphisms with Sulfolane Formation and the Efficacy of a Busulfan-Based Conditioning Regimen in Pediatric Patients Undergoing Hematopoietic Stem Cell Transplantation. Pharm. J. 2014, 14, 263–271. [Google Scholar] [CrossRef]
- Pihusch, M.; Lohse, P.; Reitberger, J.; Hiller, E.; Andreesen, R.; Kolb, H.-J.; Holler, E.; Pihusch, R. Impact of Thrombophilic Gene Mutations and Graft-versus-Host Disease on Thromboembolic Complications after Allogeneic Hematopoietic Stem-Cell Transplantation. Transplantation 2004, 78, 911–918. [Google Scholar] [CrossRef]
- Seifert, C.; Wittig, S.; Arndt, C.; Gruhn, B. Heparanase Polymorphisms: Influence on Incidence of Hepatic Sinusoidal Obstruction Syndrome in Children Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. J. Cancer Res. Clin. Oncol. 2015, 141, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Huezo-Diaz Curtis, P.; Uppugunduri, C.R.S.; Muthukumaran, J.; Rezgui, M.A.; Peters, C.; Bader, P.; Duval, M.; Bittencourt, H.; Krajinovic, M.; Ansari, M. Association of CTH Variant with Sinusoidal Obstruction Syndrome in Children Receiving Intravenous Busulfan and Cyclophosphamide before Hematopoietic Stem Cell Transplantation. Pharm. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Petrykey, K.; Rezgui, M.A.; Del Vecchio, V.; Cortyl, J.; Ralph, R.-O.; Nava, T.; Beaulieu, P.; St-Onge, P.; Jurkovic Mlakar, S.; et al. Genetic Susceptibility to Hepatic Sinusoidal Obstruction Syndrome in Pediatric Patients Undergoing Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2020, 26, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Vreuls, C.P.H.; Olde Damink, S.W.M.; Koek, G.H.; Winstanley, A.; Wisse, E.; Cloots, R.H.E.; van den Broek, M.a.J.; Dejong, C.H.C.; Bosman, F.T.; Driessen, A. Glutathione S-Transferase M1-Null Genotype as Risk Factor for SOS in Oxaliplatin-Treated Patients with Metastatic Colorectal Cancer. Br. J. Cancer 2013, 108, 676–680. [Google Scholar] [CrossRef]
- Elbahlawan, L.; McArthur, J.; Quasney, M.W.; Pei, D.; Srivastava, K.; Dahmer, M.K.; Barfield, R. Association of IL-1β -511 Polymorphism with Severe Veno-Occlusive Disease in Pediatric-Matched Allogeneic Hematopoietic Stem Cell Transplantation. J. Pediatr. Hematol. Oncol. 2012, 34, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Sucak, G.T.; Yaşar, D.G.; Yegin, Z.A.; Ergün, M.A.; Ozkurt, Z.N.; Aki, Ş.Z.; Güntekin, S. The Prognostic Role of Hemochromatosis H63D Allele in Allogeneic Hematopoietic Stem Cell Transplantation. Ann. Hematol. 2012, 91, 1281–1287. [Google Scholar] [CrossRef]
- Ansari, M.; Curtis, P.H.-D.; Uppugunduri, C.R.S.; Rezgui, M.A.; Nava, T.; Mlakar, V.; Lesne, L.; Théoret, Y.; Chalandon, Y.; Dupuis, L.L.; et al. GSTA1 Diplotypes Affect Busulfan Clearance and Toxicity in Children Undergoing Allogeneic Hematopoietic Stem Cell Transplantation: A Multicenter Study. Oncotarget 2017, 8, 90852–90867. [Google Scholar] [CrossRef] [PubMed]
- Goekkurt, E.; Stoehlmacher, J.; Stueber, C.; Wolschke, C.; Eiermann, T.; Iacobelli, S.; Zander, A.R.; Ehninger, G.; Kröger, N. Pharmacogenetic Analysis of Liver Toxicity after Busulfan/Cyclophosphamide-Based Allogeneic Hematopoietic Stem Cell Transplantation. Anticancer Res. 2007, 27, 4377–4380. [Google Scholar] [PubMed]
- Zwaveling, J.; Press, R.R.; Bredius, R.G.M.; van Derstraaten, T.R.J.H.M.; den Hartigh, J.; Bartelink, I.H.; Boelens, J.J.; Guchelaar, H.-J. Glutathione S-Transferase Polymorphisms Are Not Associated with Population Pharmacokinetic Parameters of Busulfan in Pediatric Patients. Drug Monit 2008, 30, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Orchard, P.J.; Baker, K.S.; Brundage, R.; Cao, Q.; Wang, X.; Langer, E.; Farag-El Maasah, S.; Ross, J.A.; Remmel, R.; et al. Glutathione S-Transferase A1 Genetic Variants Reduce Busulfan Clearance in Children Undergoing Hematopoietic Cell Transplantation. J. Clin. Pharm. 2008, 48, 1052–1062. [Google Scholar] [CrossRef]
- Byun, J.M.; Kim, H.-L.; Shin, D.-Y.; Koh, Y.; Yoon, S.-S.; Seong, M.-W.; Park, S.S.; Kim, J.H.; Lee, Y.-G.; Kim, I. The Impact of Methylenetetrahydrofolate Reductase C677T Polymorphism on Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation with Methotrexate Prophylaxis. PLoS ONE 2016, 11, e0163998. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.; Dodds, A.; Atkinson, K.; Szer, J.; Downs, K.; Biggs, J. The Toxicity of Busulphan and Cyclophosphamide as the Preparative Regimen for Bone Marrow Transplantation. Br. J. Haematol. 1991, 77, 529–534. [Google Scholar] [CrossRef]
- Czerwinski, M.; Gibbs, J.P.; Slattery, J.T. Busulfan Conjugation by Glutathione S-Transferases Alpha, Mu, and Pi. Drug Metab. Dispos. 1996, 24, 1015–1019. [Google Scholar]
- Coles, B.F.; Morel, F.; Rauch, C.; Huber, W.W.; Yang, M.; Teitel, C.H.; Green, B.; Lang, N.P.; Kadlubar, F.F. Effect of Polymorphism in the Human Glutathione S-Transferase A1 Promoter on Hepatic GSTA1 and GSTA2 Expression. Pharmacogenetics 2001, 11, 663–669. [Google Scholar] [CrossRef]
- Nava, T.; Rezgui, M.A.; Uppugunduri, C.R.S.; Curtis, P.H.-D.; Théoret, Y.; Duval, M.; Daudt, L.E.; Ansari, M.; Krajinovic, M.; Bittencourt, H. GSTA1 Genetic Variants and Conditioning Regimen: Missing Key Factors in Dosing Guidelines of Busulfan in Pediatric Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1918–1924. [Google Scholar] [CrossRef]
- Ten Brink, M.H.; van Bavel, T.; Swen, J.J.; Straaten, T.; Bredius, R.G.; Lankester, A.C.; Zwaveling, J.; Guchelaar, H.-J. Effect of Genetic Variants GSTA1 and CYP39A1 and Age on Busulfan Clearance in Pediatric Patients Undergoing Hematopoietic Stem Cell Transplantation. Pharmacogenomics 2013, 14, 1683–1690. [Google Scholar] [CrossRef]
- Kodidela, S.; Kumar, S.S.; Uppugunduri, C.R.S. Developmental Pattern of Hepatic Drug‑Metabolizing Enzymes in Pediatric Population and Its Role in Optimal Drug Treatment. Arch. Med. Health Sci. 2017, 5, 8. [Google Scholar]
- McCune, J.S.; Bemer, M.J.; Barrett, J.S.; Scott Baker, K.; Gamis, A.S.; Holford, N.H.G. Busulfan in Infant to Adult Hematopoietic Cell Transplant Recipients: A Population Pharmacokinetic Model for Initial and Bayesian Dose Personalization. Clin. Cancer Res. 2014, 20, 754–763. [Google Scholar] [CrossRef]
- Corbacioglu, S.; Jabbour, E.J.; Mohty, M. Risk Factors for Development of and Progression of Hepatic Veno-Occlusive Disease/Sinusoidal Obstruction Syndrome. Biol. Blood Marrow Transplant. 2019, 25, 1271–1280. [Google Scholar] [CrossRef]
- Zanger, U.M.; Klein, K. Pharmacogenetics of Cytochrome P450 2B6 (CYP2B6): Advances on Polymorphisms, Mechanisms, and Clinical Relevance. Front. Genet. 2013, 4. [Google Scholar] [CrossRef]
- Goyette, P.; Sumner, J.S.; Milos, R.; Duncan, A.M.V.; Rosenblatt, D.S.; Matthews, R.G.; Rozen, R. Human Methylenetetrahydrofolate Reductase: Isolation of CDNA, Mapping and Mutation Identification. Nat. Genet. 1994, 7, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. Homocysteine, a Risk Factor for Cardiovascular Disease. Int. J. Angiol. 1999, 8, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Storb, R.; Deeg, H.J.; Whitehead, J.; Appelbaum, F.; Beatty, P.; Bensinger, W.; Buckner, C.D.; Clift, R.; Doney, K.; Farewell, V. Methotrexate and Cyclosporine Compared with Cyclosporine Alone for Prophylaxis of Acute Graft versus Host Disease after Marrow Transplantation for Leukemia. N. Engl. J. Med. 1986, 314, 729–735. [Google Scholar] [CrossRef]
- Kato, M.; Ishimaru, S.; Seki, M.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Kakiuchi, N.; Sato, Y.; Ueno, H.; Tanaka, H.; et al. Long-Term Outcome of 6-Month Maintenance Chemotherapy for Acute Lymphoblastic Leukemia in Children. Leukemia 2017, 31, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Characteristics | n | Proportion (%) |
|---|---|---|
| Centers included | ||
| monocentric | 20 | 74.1 |
| multicentric | 6 | 22.2 |
| unclear | 1 | 3.7 |
| Location | ||
| Europe | 11 | 40.7 |
| North America | 9 | 33.3 |
| Asia | 4 | 14.8 |
| Others | 3 | 11.1 |
| Study design | ||
| cohort | 23 | 85.2 |
| prospective trial | 2 | 7.4 |
| case-control | 2 | 7.4 |
| Sample size | ||
| median, IQR (n) | 84 | 65–142 |
| 0–50 | 3 | 11.1 |
| 51–100 | 13 | 48.1 |
| 101–150 | 4 | 14.8 |
| 151–200 | 4 | 14.8 |
| 201 and more | 3 | 11.1 |
| Age group at treatment | ||
| children and adolescents only | 10 | 37 |
| children, adolescents, and adults | 11 | 40.7 |
| adults only | 6 | 22.2 |
| Treatment exposure | ||
| allogeneic HSCT, busulfan-based | 10 | 37 |
| allogeneic HSCT, various regimens | 9 | 33.3 |
| autologous and allogeneic HSCT | 4 | 14.8 |
| non-HSCT | 4 | 14.8 |
| Outcome | ||
| incidence cohort-based samples (mean %, range %) | 16.5 | 2.3–42.9 |
| (modified) Seattle criteria | 15 | 55.6 |
| Baltimore criteria | 5 | 18.5 |
| other criteria/unspecified | 7 | 25.9 |
| Association analysis | ||
| candidate gene analysis | 26 | 96.3 |
| genome/exome wide analysis | 1 | 3.7 |
| Lead Author, Journal Year | Study Design | Location | Population (Diagnoses, Age) | Exposure, Location | n (SOS/Total) | Genes/Region | Variants Investigated | OR/ RR (CI) | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| Duggan C, et al. Bone Marrow Transplant. 1999. [22] | Candidate-gene; case-control | St James’s Hospital and Trinity College Dublin, Ireland | Unclear diagnoses, median age 29 years (range 4–55) | AlloHSCT and autoHSCT with various regimens (Bu, Cy, Mel, TBI, others) | 22/287 (7.7%), genotyped: 15/51 (29.4%) | F2 | rs1799963(GA vs. GG) | - | p = 0.05 |
| F5 | rs6025(GG vs. AG/AA) | - | p = 0.05 | ||||||
| Pihusch M, et al. Transplantation. 2004 [44] | Candidate-gene; cohort | José-Carreras transplantation unit Munich, Germany | Various malignant and non-malignant diagnoses; median age 43 years (range 14–62) | AlloHSCT with various regimens (Bu, Cy, Mel, TBI, others) | 3/89 (3.4%) | F2 | rs1799963(G > A) | “no effect” | - |
| F5 | rs6025(G > A) | “no effect” | - | ||||||
| MTHFR | rs1801133(C > T) | “no effect” | - | ||||||
| ITGB3 | rs591(C > T) | “no effect” | - | ||||||
| FGB | rs1800790(G > A) | “no effect” | - | ||||||
| SERPINE1 | rs1799889(4G allele) | (83.3% vs. 55.1%) | NS | ||||||
| ACE | rs1799752(D allele) | “no effect” | - | ||||||
| Srivastava A, et al. Blood. 2004 [42] | Candidate-gene; cohort | Hôpital Robert Debré, Paris, France | Beta-thalassemia major; median age 6 years (range 2–16) | Busulfan–cyclophosphamide-based alloHSCT | 33/114 (28.9%) | GSTM1 | “null genotype” ‡ | OR 4.3 (1.5–12.5) † | p = 0.008 † |
| GSTT1 | “null genotype”‡ | OR 0.6 (0.2–1.9) † | p = 0.4 † | ||||||
| Kallianpur AR et al. Bone Marrow Transplant. 2005 [35] | Candidate-gene; cohort | Multicentric, two centers in Nashville, Tennessee, USA | Various hematological and solid neoplasms; mean age 44 years (range 19–64) | AlloHSCT and autoHSCT with various regimens (Bu, Cy, TBI, others) | 30/166 (18.1%) | HFE | rs1800562(A > G) | RR 3.7 (1.2–12.1); RR 1.7 (0.4–6.8) for heterozygotes; RR 8.6 (1.5–48.5) for homozygotes † | p = 0.01 † |
| CPS1 | rs7422339(CC vs. AC/AA) | - | p = 0.038 | ||||||
| Elmaagacli AH, et al. Bone Marrow Transplant. 2007 [34] | Candidate-gene; cohort | University Hospital of Essen, Germany | Various hematological neoplasms incl. lymphomas; median age 41 years (range 17–67) | AlloHSCT with various regimens (Bu, Cy, TBI, others) | 20/286 (7%) | CYP2C19 | Poor vs. intermediate/extensive metabolizers (rs4244285(AA vs. AG/GG) rs4986893(AA vs. AG/GG)) | - | NS |
| Goekkurt E, et al. Anticancer Res. 2007 [52] | Candidate-gene; cohort | University Hospital Hamburg, Germany | Various hematological malignancies and non-malignant diagnoses; median age 39.5 years (range 16–59) | Busulfan–cyclophosphamide-based alloHSCT | 36/84 (42.9%) | GSTA1 | * B vs. * A haplotypes | - | NS |
| GSTM1 | “null genotype”‡ | - | NS | ||||||
| GSTP1 | rs1695(A > G) | - | NS | ||||||
| GSTT1 | “null genotype”‡ | - | NS | ||||||
| MTHFR | rs1801133(C > T) | - | NS | ||||||
| rs1801131(A > C) | OR 9.4 (1.1–81.9) † | p = 0.048 † | |||||||
| Kim I, et al. Annals of Hematol. 2007 [38] | Candidate-gene; cohort | Seoul National University College of Medicine, South Korea | Hematological malignancies and aplastic anemia; median age 36 year (range 16–52) | AlloHSCT with various regimens (Bu, Cy, TBI) | 11/72 (15.3%) | MTHFR | rs1801133(C > T) | - | p = 0.4 |
| rs1801131(A > C) | - | p = 0.48 | |||||||
| Lee KH, et al. Haematologica. 2007 [39] | Candidate-gene; cohort | Seoul National University Hospital, South Korea | Hematological malignancies incl. lymphomas and aplastic anemia; median age 40 years (range 16–70) | AlloHSCT with various regimens (Bu, Cy, Mel, TBI, others) from HLA-matched sibling donors | 19/152 (12.5%) | P2RX7 | rs3751143(A > C) | - | p = 0.78 |
| Zwaveling J, et al. Therapeut Drug Monitor. 2008 [53] | Candidate-gene; cohort | Multicentric, pediatric Leiden and Utrecht University Medical Centers, Netherlands | Hematological malignancies and non-malignant diagnoses; median age 5 years (range 0.2–23) | Busulfan-based alloHSCT with various other agents (Cy, Mel, others) | 15/77 (19.5%) | GSTA1 | rs3957357(C > T) | - | - |
| GSTM1 | “null genotype” ‡ | - | p = 0.07 | ||||||
| GSTP1 | rs1695(A > G) | - | - | ||||||
| GSTT1 | “null genotype” ‡ | - | - | ||||||
| Johnson L, et al. J Clin Pharmacol. 2008 [54] | Candidate-gene; cohort | University of Minnesota, USA | Malignant and nonmalignant diagnoses; median age 5.6 years (range 0.1–18.3) | Busulfan-based alloHSCT with various other agents (Cy, others) | 3/29 (10.3%) | GSTA1 | * B vs. * A haplotypes | - | NS |
| GSTM1 | “null genotype” ‡ | - | NS | ||||||
| GSTP1 | rs1695(A > G) | - | NS | ||||||
| rs1138272(C > T) | - | NS | |||||||
| Rocha V, et al. Leukemia. 2009 [40] | Candidate-gene; cohort | Hôpital Saint Louis, Paris, France | Acute and chronic leukemia; median age 35 years (range 3–56) | AlloHSCT with various regimens (Bu, Cy, Mel, TBI, others) from HLA-matched sibling donors | 15/107 (14%) | CYP2B6 | * 2A haplotype | - | NA |
| * 4 haplotype | - | NA | |||||||
| * 5 haplotype | - | NA | |||||||
| * 6 haplotype | OR 3.49 (1.12–10.88) † | p = 0.03 † | |||||||
| GSTM1 | “null genotype” ‡ | - | NA | ||||||
| GSTP1 | rs1695(AA vs. AG/GG) | NA | |||||||
| GSTT1 | “null genotype” ‡ | - | NA | ||||||
| ABCB1 | rs1045642(CC vs. CT/TT) | - | NA | ||||||
| MTHFR | rs1801133(CC vs. CT/TT) | NA | |||||||
| VDR | Apal (rs7975232) | - | NA | ||||||
| BsmI (rs1544410) | NA | ||||||||
| TaqI (rs731236) | - | NA | |||||||
| Elbahlawan L, et al. J Ped Hem Oncol. 2012 [49] | Candidate-gene; cohort | St Jude Children’s Research Hospital, USA | Malignant and non-malignant diagnoses; median age 10.1 years (range 1–19.6) | AlloHSCT with various regimens (Bu, Cy, TBI, others) from HLA-matched donors | 5/76 (6.6%) | IL1B | rs16944(A > G) | - | p = 0.18 |
| Sucak GT, et al. Ann Hematology. 2012 [50] | Candidate-gene; cohort | Gazi University, Ankara, Turkey | Malignant and non-malignant diagnoses; median age 27.5 years (range 16–64) | AlloHSCT with various regimens (Bu, Mel, TBI, others) | 22/102 (21.6%) | HFE | rs1799945(C > G) | - | p > 0.05 |
| Krivoy N, et al. Curr Drug Safety. 2012 [37] | Candidate-gene; cohort | Technion-Israel Institute of Technology; Haifa, Israel | Acute myeloid leukemia; median age 39.2 years (SD 12.3) | Busulfan–cyclophosphamide-based autoHSCT and alloHSCT | 8/63 (12.7%) | ABCB1 | rs1045642(C > T) | - | NS |
| rs2032582(G > T/A) | - | NS | |||||||
| GSTA1 | rs3957357(C > T) | - | NS | ||||||
| GSTM1 | “null genotype” ‡ | - | NS | ||||||
| GSTP1 | rs1695(A > G) | - | p = 0.05 | ||||||
| GSTT1 | “null genotype” ‡ | - | NS | ||||||
| Uppugunduri CRS, et al. Pharmacogenom J. 2014 [43] | Candidate-gene; cohort | CHU Sainte-Justine, Montreal, Canada | Malignant and non-malignant diagnoses; median age 6.9 years (range 0.1–19.9) | Busulfan–based alloHSCT with various other agents (Cy, Mel, TBI, others) | 8/66 (12.1) | CYP2B6 | rs3211371(C > T) | - | NS |
| rs3745274(G > T) | - | NS | |||||||
| CYP2C19 | rs4244285(G > A) | - | NS | ||||||
| rs12248560(C > T) | - | NS | |||||||
| CYP2C9 | rs1799853(C > T) | - | NS | ||||||
| rs1057910(G > A) | - | NS | |||||||
| FMO3 | rs2266780(A > G) | - | NS | ||||||
| rs2266782(G > A) | - | NS | |||||||
| rs1736557(A > G) | - | NS | |||||||
| Efrati E, et al. Bone Marrow Transplant. 2014 [33] | Candidate-gene; cohort | Technion-Israel Institute of Technology; Haifa, Israel | Acute myeloid leukemia; adult cohort | Busulfan–cyclophosphamide-based alloHSCT (with TBI in one) | 9/62 (15%) | MTHFR | rs1801133(CC vs. CT/TT) | - | p = 0.0096 |
| rs1801131(CC vs. AC/AA) | - | p = 0.0002 | |||||||
| Seifert C, et al. J. Cancer Res. Clin. Oncol. 2015 [45] | Candidate-gene; cohort | Jena University Hospital, Germany | Malignant and non-malignant diagnoses; median age 14 years, (range 0–29) | AlloHSCT with various regimens (Bu, Cy, Mel, TBI) | 12/160 (7.5%) | HPSE | rs4693608(AA vs. AG/GG) | - | p = 0.038 |
| rs4364254(TT vs. TC/CC) | - | p = 0.004 | |||||||
| rs4693608(AA)and rs4364254(TT) † | 4.06 (1.14–14.4) † | p = 0.03 † | |||||||
| Ansari M, et al. Bone Marrow Transplant. 2016 [41] | Candidate-gene; cohort | San Raffaele Institute, Milan, Italy | Thalassemia intermedia (20.5%) and thalassemia major (79.5%); median age 8 years (range 1.5–17) | Busulfan–cyclophosphamide-based alloHSCT from HLA-matched sibling donors | 1/44 (2.3%) | GSTA1 | * B vs. * A haplotypes using rs3957357(C > T) | - | NS |
| GSTM 1 | “null genotype” ‡ | - | NS | ||||||
| Byun JM, et al. PloS One. 2016 [55] | Candidate-gene; cohort | Seoul National University Hospital, South Korea | Hematological malignancies incl. lymphomas and aplastic anemia; median age 37.8 years (SD 12.5) | AlloHSCT with various regimens (not further specified) | 10/177 (5.6%) | MTHFR | rs1801133(TT vs. CT/CC) | - | p = 0.234 |
| Huezo-Diaz Curtis p, et al. Pharmacogenomics J. 2016 [46] | Candidate-gene; cohort | CHU Sainte-Justine, Montreal, Canada | Malignant and non-malignant diagnoses; median age 6.4 years (range 0.1–19.9) | Busulfan-based alloHSCT with various other agents (Cy, Mel, others) | 9/76 (11.8%) | CTH | rs1021737(TT vs. GT/GG) | OR 10.6 (2.2–51.5) | p = 0.003 |
| rs648743(C > T) | - | NS | |||||||
| GSTA1 | * B* B vs. * A* B/* A* A haplotypes | OR 10.9 (2.3–51.3) | p = 0.007 | ||||||
| Ansari M, et al. Oncotarget. 2017; [51] → includes all patients from: [29] and [30] | Candidate-gene; cohort | Multicentric: Geneva, Leiden, Montreal, Paris, Toronto | Malignant and non-malignant diagnoses; median age 5.8 years (range 0.1–19.9) | Busulfan-based alloHSCT with various other agents (Cy, Mel, others) | 14/138 (10%) | GSTA1 | Slow metabolizer haplotypes (group IV) | OR 9.0 (2.6–31) † | p = 0.001 † |
| GSTM1 | “null genotype” ‡ | - | NA | ||||||
| GSTP1 | rs1695(A > G) | - | NA | ||||||
| rs1138272(C > T) | - | NA | |||||||
| Ansari M, et al. Biology of Blood and Marrow Transplantation. 2020 [47] | Exome-wide association analysis with replication in an independent sample; cohort | Discovery cohort: CHU Sainte-Justine, Montreal, Canada; replication cohort: multicentric | Malignant and non-malignant diagnoses; median age discovery: 7.4 years (range 0–23.5); replication: 4.7 years (range 0–21) | Busulfan-based alloHSCT with various other agents (Cy, Mel, others) | Discovery: 12/87 (13.8%); replication: 27/182 (14.8%) | UGT2B10 | rs17146905A > G | OR 8.4 (3.0–23.9) | p = 7 × 10−6 (replication p = 0.0004 †) |
| KIAA1715 = LNPK | rs2289971T > C | OR 10.2 (3.3–31.9) | p = 3 × 10−6 (replication p = 0.05 †) | ||||||
| BHLHE22 | rs16931326G > A | OR 8.9 (2.9–26.9) | p = 1.1 × 10−5 (replication p > 0.05 §) | ||||||
| HADH | rs17511319A > G | OR 30.5 (5.9–158.6) | p = 1.2 × 10−5 (replication p = 0.05) | ||||||
| ZNF608 | rs75323508 C > T | OR 9.9 (3.0–32.8) | p = 1.3 × 10−5 (replication p = 0.4) | ||||||
| AMPH | rs2810T > C | OR 8.9 (2.9–26.9) | p = 1.1 × 10−5 (replication p = 0.9) | ||||||
| FAT3 | rs11823754G > T | OR 10.7 (3.6–31.7) | p = 8.3 × 10−7 (replication p = 1.0) | ||||||
| AGPAT3 | rs11537798A > G | OR 9.9 (3.0–32.8 | p = 1.3 × 10−5 (replication p = 0.1) | ||||||
| GSTA1 | Slow metabolizer haplotypes (group IV) | OR 3.1 (1.2–8.0) in replication cohort † | replication cohort only: p = 0.02 † | ||||||
| Terakura S, et al. Int J Hematol. 2020 [36] | Candidate-gene; cohort | Nagoya University Hospital, Japan | Hematological malignancies incl. lymphomas; median age 38 years (21–67) | Busulfan–cyclophosphamide based autoHSCT and alloHSCT | 8/55 (14.5%) | CYP2B6 | rs3745274(G > T) | - | NS |
| rs2279344(A > G) | - | NS | |||||||
| rs2099361(A > C) | - | NS | |||||||
| rs8100458(C > T) | - | NS | |||||||
| rs2014141(A > G) | - | NS | |||||||
| CYP2C9 | rs1799853 | - | NS | ||||||
| rs1057910(A > C) | - | NS | |||||||
| CYP2C19 | rs4986893 (G > A) | - | NS | ||||||
| rs4244285(G > A) | - | NS | |||||||
| GSTA1 | * B vs. * A haplotype (rs4715326) | - | NS | ||||||
| GSTM1 | “null genotype” ‡ | - | NS | ||||||
| GSTO1 | rs4925(A > C) | - | NS | ||||||
| rs11191972(C > T) | - | NS | |||||||
| GSTO2 | rs156697(A > G) | - | NS | ||||||
| rs2297235(A > G) | - | NS | |||||||
| GSTP1 | rs1695(A > G) | - | NS | ||||||
| rs614080((A > G) | - | NS | |||||||
| GSTT1 | “null genotype” ‡ | - | NS | ||||||
| GSTZ1 | rs2270423(A > G) | - | NS |
| Lead Author, Journal Year | Study Design | Location | Population (Diagnoses, Age) | Exposure, Location | n (SOS/Total) | Genes/Region | Variants Investigated | OR (CI) | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| Aplenc R, et al. Acta Haematologica. 2003 [32] | Candidate-gene; case-control | University of Washington Medical Center, Seattle, USA | Relapsed AML; mean age 45.4 years | Gemtuzumab for relapsed disease after HSCT (SOS not primarily associated with HSCT) | 11/21 (52%) Genotyped: 9/18 (50%) | GSTM1 | “null genotype” ‡ | - | NS |
| GSTT1 | “null genotype” ‡ | - | NS | ||||||
| GSTP1 | *B haplotype | OR 4 (NA) | NS | ||||||
| *C haplotype | - | NS | |||||||
| NQ01 | *2 haplotype | - | NS | ||||||
| Lennard L, et al. Clin. Pharmacol. Ther. 2006 [12] | Candidate-gene; case-control based on prospective trial | Multicentric, USA | Acute lymphoblastic leukemia; median age 4 years (range 1–16) | Treatment according to protocols CCG-ALL97 (n = 33/393 with SOS, 8%) and CCG-ALL99 (n = 49/355 with SOS, 14%) | 50/203 (24.6%) | TPMT | *3A/*3C haplotypes | - | p = 0.11 |
| Vreuls CPH, et al. Brit J Cancer. 2013 [48] | Candidate-gene; cohort | Maastricht University Medical Centre, NL | Patients with metastatic colorectal cancer; mean age 62 years (range 40–81) | Initial partial hepatic resection and treatment with oxaliplatin | 32/55 (58%) | GSTM1 | “null genotype” ‡ | - | p = 0.026 † |
| GSTT1 | “null genotype” ‡ | - | NS | ||||||
| Wray L, et al. Pediatr Blood Cancer. 2014 [13] | Candidate-gene; prospective trial | Children’s Hospital of Philadelphia, USA | Acute lymphoblastic leukemia; pediatric patients (range 1–10 years) | Treatment according to protocol CCG-1952 | 79/351 (22.5%) | TPMT | *3A haplotype | OR 0.7 (0.3–1.6) † | NS † |
| *3B haplotype | OR 1.0 (0.4–2.6) † | NS † | |||||||
| *3C haplotype | OR 0.7 (0.2–1.8) † | NS † | |||||||
| MTHFR | rs1801133(CC vs.CT/TT) | OR 0.9 (0.3–2.4) † | NS † | ||||||
| rs1801131(CC vs.AC/AA) | OR 1.4(0.5–3.8) † | NS † |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waespe, N.; Strebel, S.; Jurkovic Mlakar, S.; Krajinovic, M.; Kuehni, C.E.; Nava, T.; Ansari, M. Genetic Predictors for Sinusoidal Obstruction Syndrome—A Systematic Review. J. Pers. Med. 2021, 11, 347. https://doi.org/10.3390/jpm11050347
Waespe N, Strebel S, Jurkovic Mlakar S, Krajinovic M, Kuehni CE, Nava T, Ansari M. Genetic Predictors for Sinusoidal Obstruction Syndrome—A Systematic Review. Journal of Personalized Medicine. 2021; 11(5):347. https://doi.org/10.3390/jpm11050347
Chicago/Turabian StyleWaespe, Nicolas, Sven Strebel, Simona Jurkovic Mlakar, Maja Krajinovic, Claudia Elisabeth Kuehni, Tiago Nava, and Marc Ansari. 2021. "Genetic Predictors for Sinusoidal Obstruction Syndrome—A Systematic Review" Journal of Personalized Medicine 11, no. 5: 347. https://doi.org/10.3390/jpm11050347
APA StyleWaespe, N., Strebel, S., Jurkovic Mlakar, S., Krajinovic, M., Kuehni, C. E., Nava, T., & Ansari, M. (2021). Genetic Predictors for Sinusoidal Obstruction Syndrome—A Systematic Review. Journal of Personalized Medicine, 11(5), 347. https://doi.org/10.3390/jpm11050347