
The Quandary of DNA-Based Treatment Assessment in De Novo Metastatic Prostate Cancer in the Era of Precision Oncology


 ,
,  and
and 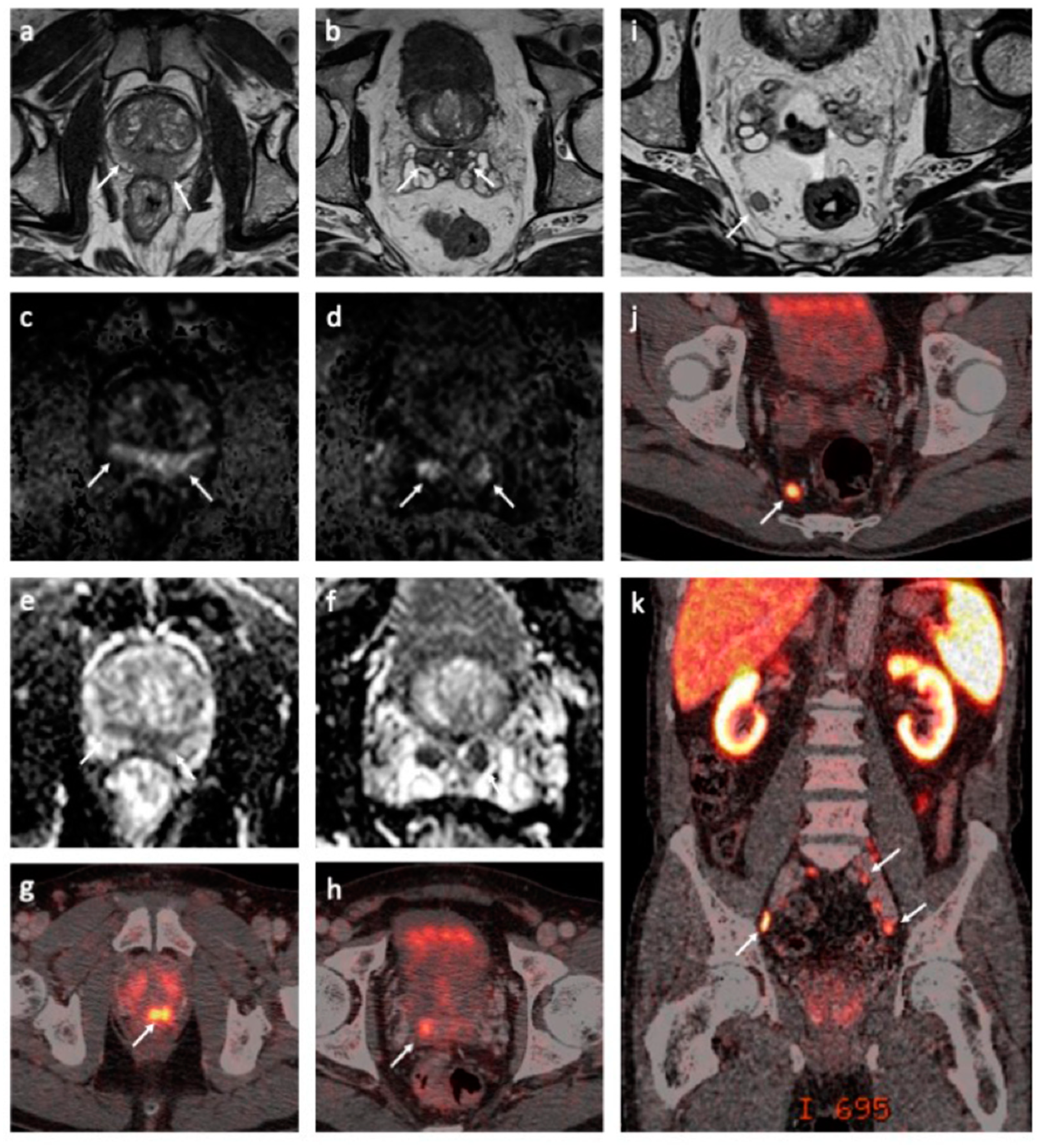{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Case History
3. Materials and Methods
3.1. DNA and RNA Extraction
3.2. Whole-Exome Sequencing Analysis
3.3. RNA Sequencing and Analysis
3.4. Immunohistochemistry
4. Results
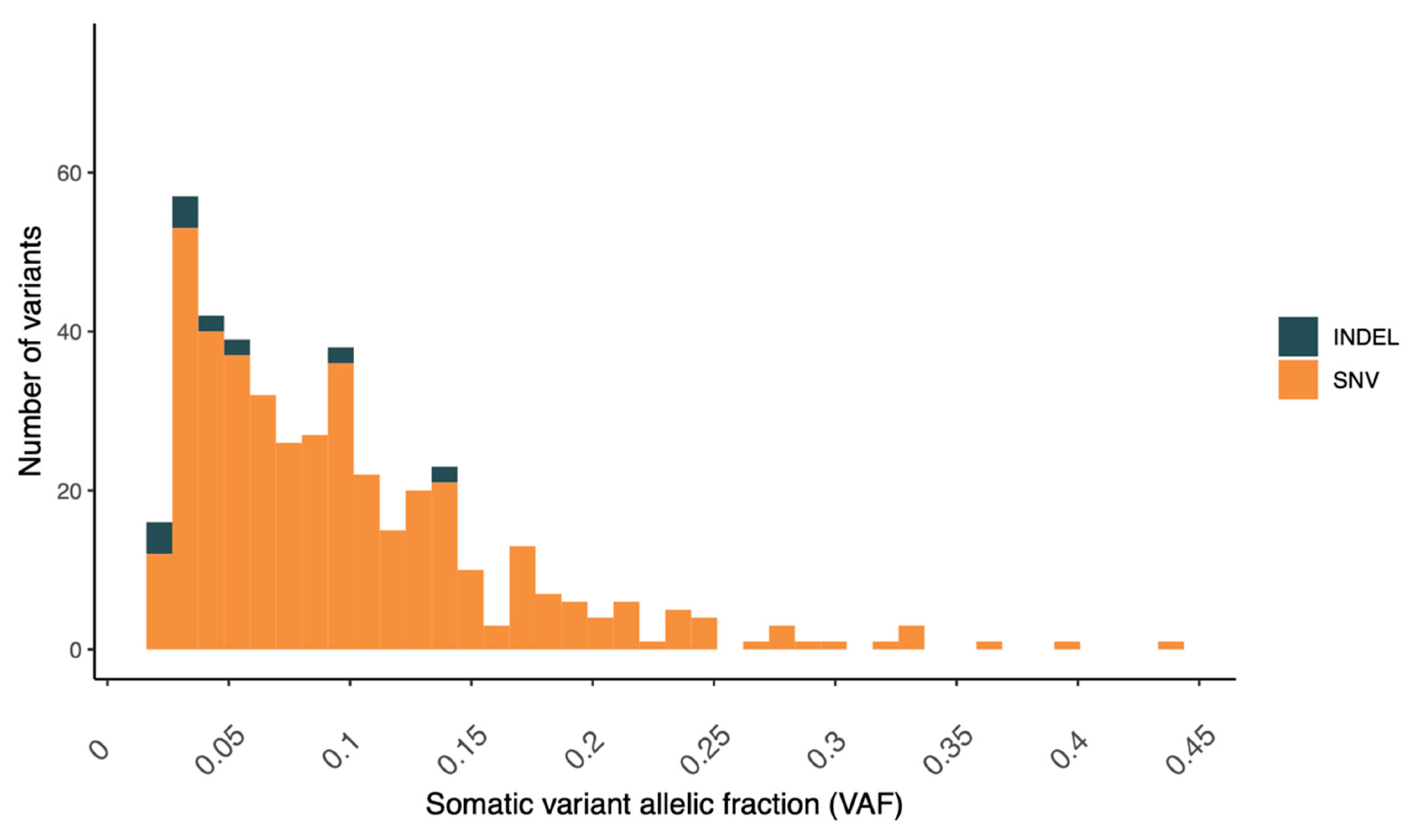
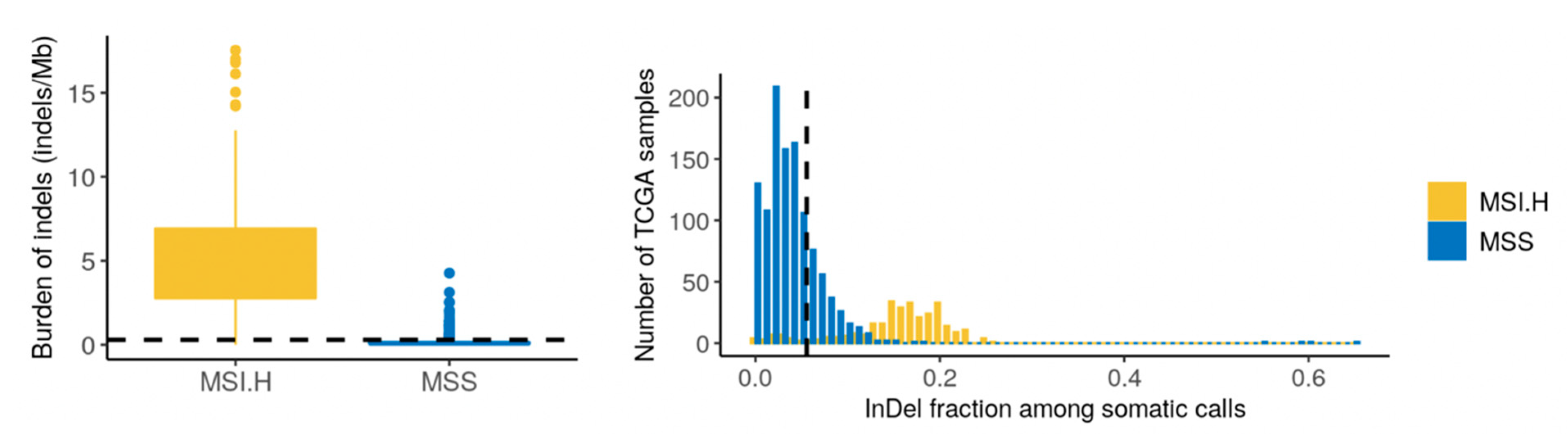
4.1. Genomic Findings—Tumor
4.2. Somatic Variants of Potential Clinical Relevance
4.2.1. PTEN Inframe Deletion—p.His196_Ile203del
4.2.2. ATM Missense Variant—p.Arg1575His
4.2.3. CREBBP Missense Variant—p.Trp1718Gly
4.3. Germline Findings—Blood
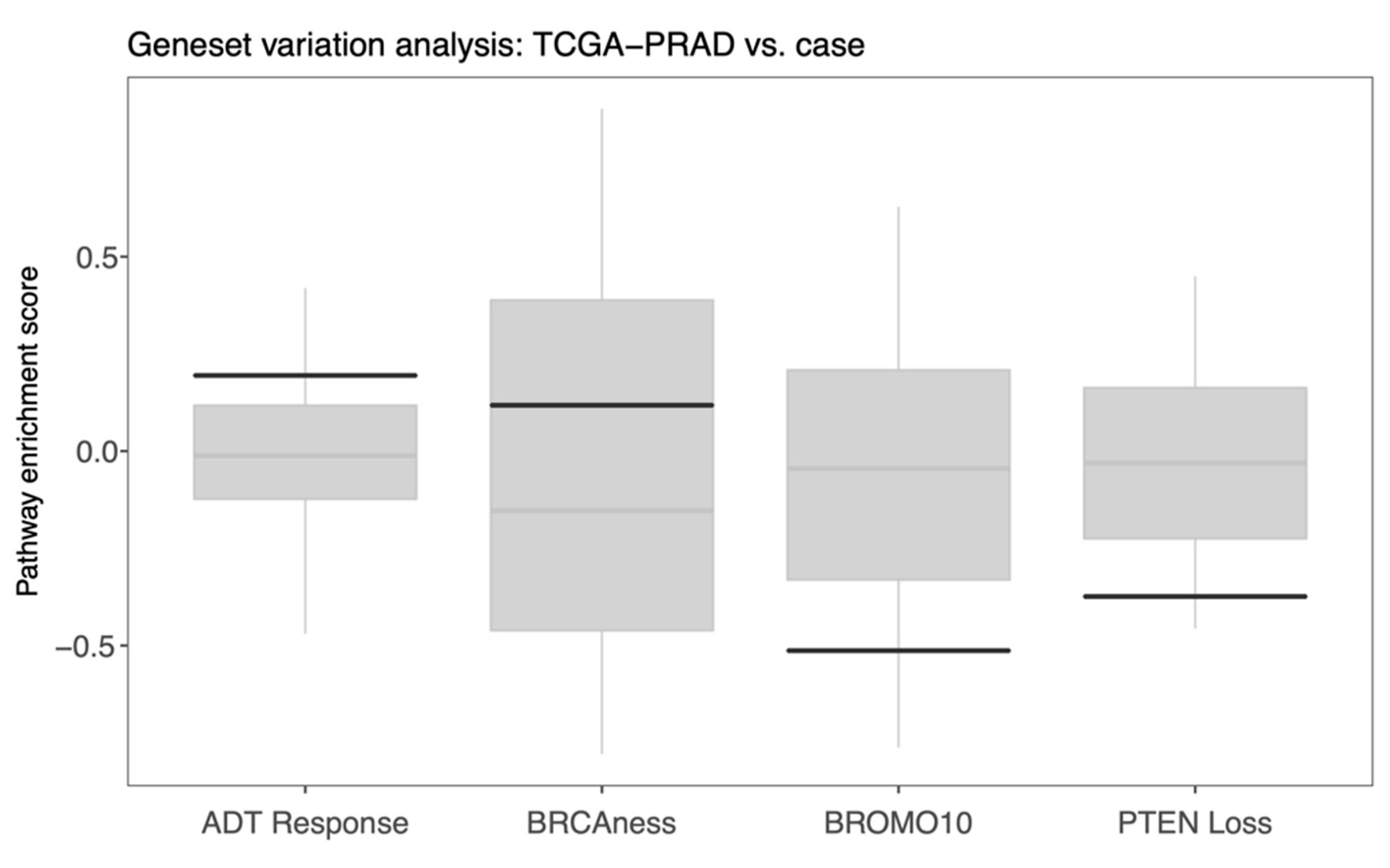
4.4. Transcriptomic Findings—Tumor
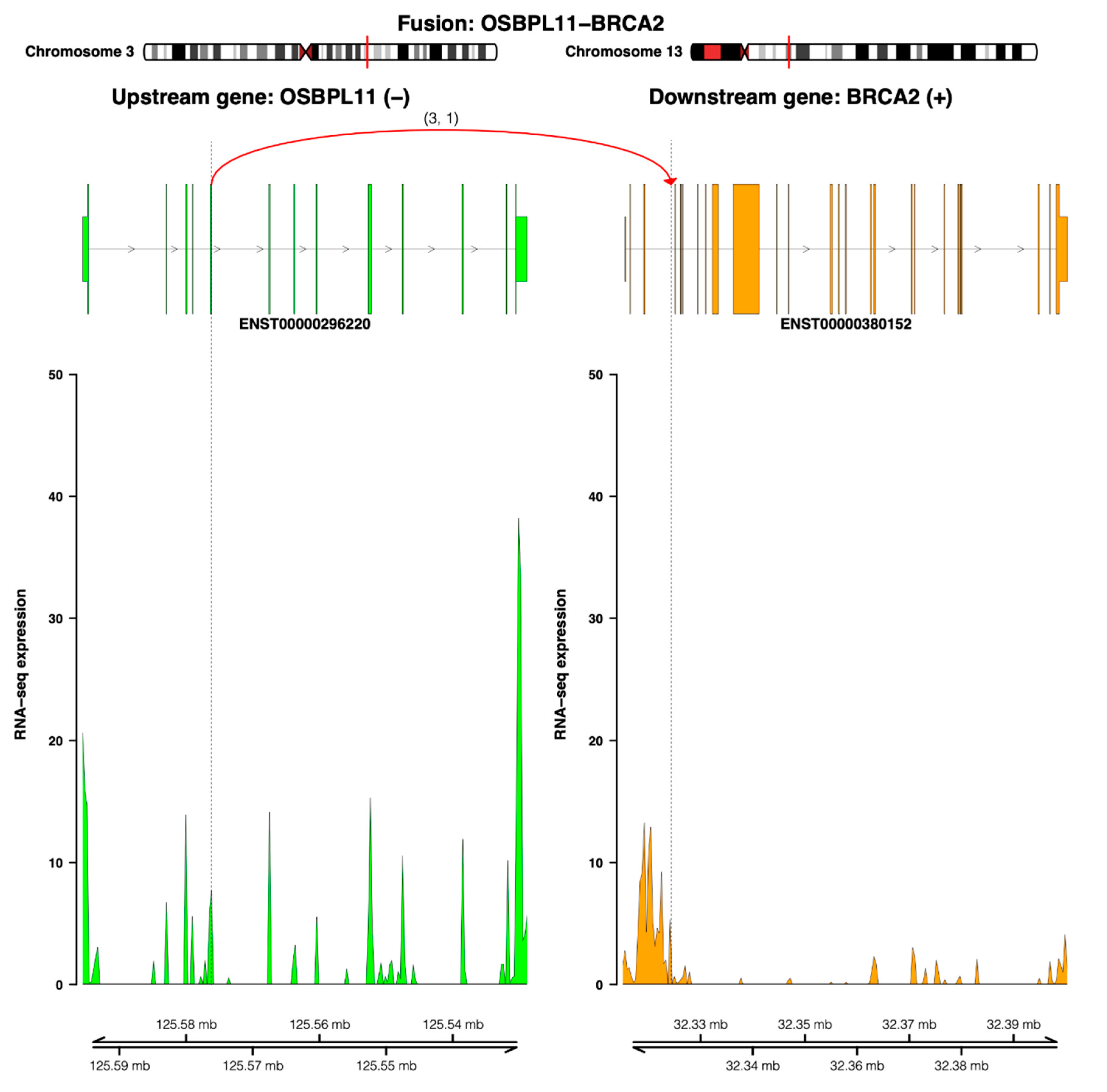
4.5. Fusion Transcript Findings—Tumor
5. Discussion
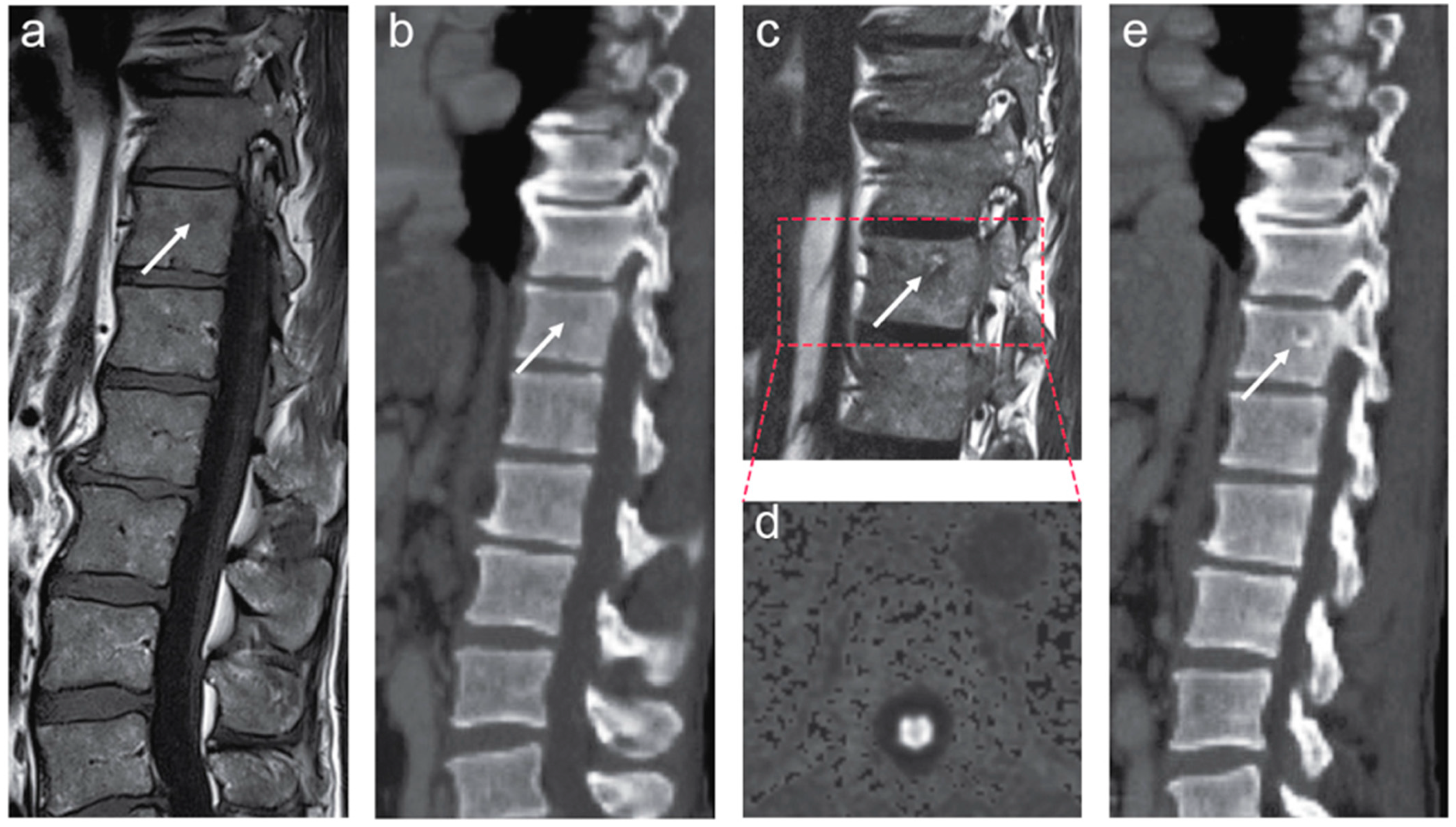
5.1. Pathology Findings
5.2. MRI Findings
5.3. Genomic and Transcriptomic Findings
5.4. Specific Treatment Considerations
5.4.1. PARP Inhibitors
5.4.2. Targeting Consequences of Loss of PTEN Activity
5.4.3. HDAC Inhibitors
5.4.4. Immune Checkpoint Inhibitors
5.4.5. Current Preferred Considerations for the Management of the Patient
- Intraductal carcinoma with grade group 5 should encourage participation in clinical trials.
- Continuous PTEN deletion as the biomarker-based monitoring of treatment efficacy in liquid biopsies, which could define a switch into therapies targeting the mTOR/PI3K/AKT axis.
- Treatment with PARPi in combination with an AR blockade.
- Immune modulation.
- Test of the ATM variant in preclinical models for the response to a treatment with PARPi and AKTi.
- Stereotactic radiotherapy to a single thoracic lesion.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garraway, L.A. Genomics-Driven Oncology: Framework for an Emerging Paradigm. J. Clin. Oncol. 2013, 31, 1806–1814. [Google Scholar] [CrossRef]
- Gagan, J.; Van Allen, E.M. Next-generation sequencing to guide cancer therapy. Genome Med. 2015, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.J.; Chen, Y.-H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.-N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- Parker, C.C.; James, N.D.; Brawley, C.D.; Clarke, N.W.; Hoyle, A.P.; Ali, A.; Ritchie, A.W.S.; Attard, G.; Chowdhury, S.; Cross, W.; et al. Radiotherapy to the primary tumour for newly diagnosed, metastatic prostate cancer (STAMPEDE): A randomised controlled phase 3 trial. Lancet 2018, 392, 2353–2366. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Shaukat, F.; Velho, P.I.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.R.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Saunders, C.T.; Wong, W.S.W.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef]
- Shen, R.; Seshan, V.E. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016, 44, e131. [Google Scholar] [CrossRef]
- Nakken, S.; Fournous, G.; Vodák, D.; Aasheim, L.B.; Myklebost, O.; Hovig, E. Personal Cancer Genome Reporter: Variant interpretation report for precision oncology. Bioinformatics 2018, 34, 1778–1780. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Nakken, S.; Saveliev, V.; Hofmann, O.; Møller, P.; Myklebost, O.; Hovig, E. Cancer Predisposition Sequencing Reporter (CPSR): A flexible variant report engine for germline screening in cancer. Cold Spring Harbor Lab. 2019, 846089. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-T.; Choi, Y.-L.; Yun, J.W.; Kim, N.K.D.; Kim, S.-Y.; Jeon, H.J.; Nam, J.-Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat. Commun. 2017, 8, 1377. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Bose, R. Genomic Alteration Burden in Advanced Prostate Cancer and Therapeutic Implications. Front. Oncol. 2019, 9, 1287. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Lee, S.; Park, W.-Y.; Kim, T.-M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, T.; Tiezzi, D.G.; Squire, J.A. Distinct subtypes of genomic PTEN deletion size influence the landscape of aneuploidy and outcome in prostate cancer. Mol. Cytogenet. 2018, 11, 1. [Google Scholar] [CrossRef]
- Burdak-Rothkamm, S.; Mansour, W.Y.; Rothkamm, K. DNA Damage Repair Deficiency in Prostate Cancer. Trends Cancer 2020, 6, 974–984. [Google Scholar] [CrossRef]
- Na, R.; Zheng, S.L.; Han, M.; Yu, H.; Jiang, D.; Shah, S.; Ewing, C.M.; Zhang, L.; Novakovic, K.; Petkewicz, J.; et al. Germline Mutations in ATM and BRCA1/2 Distinguish Risk for Lethal and Indolent Prostate Cancer and are Associated with Early Age at Death. Eur. Urol. 2017, 71, 740–747. [Google Scholar] [CrossRef]
- Lima, Z.S.; Ghadamzadeh, M.; Arashloo, F.T.; Amjad, G.; Ebadi, M.R.; Younesi, L. Recent advances of therapeutic targets based on the molecular signature in breast cancer: Genetic mutations and implications for current treatment paradigms. J. Hematol. Oncol. 2019, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, A.I.; Banerjee, T.; Wingo, M.; Duncan, K.; Ning, J.; Rodrigues, F.M.; Huang, K.-L.; Lee, C.; Chen, F.; Ding, L.; et al. Functional analysis of BARD1 missense variants in homology-directed repair and damage sensitivity. PLoS Genetics 2019, 15, e1008049. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Doultsinos, D.; Mills, I.G. Derivation and Application of Molecular Signatures to Prostate Cancer: Opportunities and Challenges. Cancers 2021, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor–induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Karnes, R.J.; Sharma, V.; Choeurng, V.; Ashab, H.A.-D.; Erho, N.; Alshalalfa, M.; Trock, B.; Ross, A.; Yousefi, K.; Tsai, H.; et al. Development and Validation of a Prostate Cancer Genomic Signature that Predicts Early ADT Treatment Response Following Radical Prostatectomy. Clin. Cancer Res. 2018, 24, 3908–3916. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytölä, V.; Itkonen, H.M.; Coleman, I.M.; Vodák, D.; Sjöblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Saal, L.H.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.-B.; Maurer, M.; Koujak, S.; Ferrando, A.A.; Malmström, P.; Memeo, L.; et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Li, X.; Tucker, M.; Li, S.; Mu, X.J.; Eng, K.W.; Sboner, A.; Rubin, M.; Gerstein, M. Molecular medicine tumor board: Whole-genome sequencing to inform on personalized medicine for a man with advanced prostate cancer. Prostate Cancer Prostatic Dis. 2021. [Google Scholar] [CrossRef]
- Kato, M.; Hirakawa, A.; Kobayashi, Y.; Yamamoto, A.; Ishida, R.; Kamihira, O.; Sano, T.; Majima, T.; Ishida, S.; Funahashi, Y.; et al. Response of intraductal carcinoma of the prostate to androgen deprivation therapy predicts prostate cancer prognosis in radical prostatectomy patients. Prostate 2020, 80, 284–290. [Google Scholar] [CrossRef]
- Deek, M.P.; Taparra, K.; Phillips, R.; Velho, P.I.; Gao, R.W.; Deville, C.; Song, D.Y.; Greco, S.; Carducci, M.; Eisenberger, M.; et al. Metastasis-directed Therapy Prolongs Efficacy of Systemic Therapy and Improves Clinical Outcomes in Oligoprogressive Castration-resistant Prostate Cancer. Eur. Urol. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Switlyk, M.D.; Salberg, U.B.; Geier, O.M.; Vlatkovic, L.; Lilleby, W.; Lyng, H.; Seierstad, T. PTEN Expression in Prostate Cancer: Relationship with Clinicopathologic Features and Multiparametric MRI Findings. Am. J. Roentgenol. 2019, 212, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Hanousková, L.; Řezáč, J.; Veselý, Š.; Průša, R.; Kotaška, K. Diagnostic Benefits of Mindin as a Prostate Cancer Biomarker: Dijagnostičke Prednosti Mindina Kao Biomarkera Raka Prostate. J. Med. Biochem. 2020, 39, 108–111. [Google Scholar] [PubMed]
- Terada, N.; Akamatsu, S.; Kobayashi, T.; Inoue, T.; Ogawa, O.; Antonarakis, E.S. Prognostic and Predictive Biomarkers in Prostate Cancer: Latest Evidence and Clinical Implications. Ther. Adv. Med. Oncol. 2017, 9, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Davies, A.A.; Masson, J.-Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in Control of the RAD51 Recombination and DNA Repair Protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Wang, J.; Freeman, M.L.; Kirschner, A.N. Radiosensitization by enzalutamide for human prostate cancer is mediated through the DNA damage repair pathway. PLoS ONE 2019, 14, e0214670. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Gomella, L.G.; Petrylak, D.P. When and How to Use PARP Inhibitors in Prostate Cancer: A Systematic Review of the Literature with an Update on On-Going Trials. Eur. Urol. Oncol. 2020, 3, 594–611. [Google Scholar] [CrossRef] [PubMed]
- Jenzer, M.; Keß, P.; Nientiedt, C.; Endris, V.; Kippenberger, M.; Leichsenring, J.; Stögbauer, F.; Haimes, J.; Mishkin, S.; Kudlow, B.; et al. The BRCA2 mutation status shapes the immune phenotype of prostate cancer. Cancer Immunol. Immunother. 2019, 68, 1621–1633. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis from the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.J.D.M.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Rodrigues, D.N.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef]
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Fléchon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e3. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Subudhi, S.K.; Vence, L.; Zhao, H.; Blando, J.; Yadav, S.S.; Xiong, Q.; Reuben, A.; Aparicio, A.; Corn, P.G.; Chapin, B.F.; et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Mercader, M.; Bodner, B.K.; Moser, M.T.; Kwon, P.S.; Park, E.S.; Manecke, R.G.; Ellis, T.M.; Wojcik, E.M.; Yang, D.; Flanigan, R.C.; et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 14565–14570. [Google Scholar] [CrossRef]
- Drake, C.G.; Doody, A.D.H.; Mihalyo, M.A.; Huang, C.-T.; Kelleher, E.; Ravi, S.; Hipkiss, E.L.; Flies, D.B.; Kennedy, E.P.; Long, M.; et al. Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell 2005, 7, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Ebelt, K.; Babaryka, G.; Frankenberger, B.; Stief, C.G.; Eisenmenger, W.; Kirchner, T.; Schendel, D.J.; Noessner, E. Prostate cancer lesions are surrounded by FOXP3+, PD-1+ and B7-H1+ lymphocyte clusters. Eur. J. Cancer 2009, 45, 1664–1672. [Google Scholar] [CrossRef]
- Lilleby, W.; Gaudernack, G.; Brunsvig, P.F.; Vlatkovic, L.; Schulz, M.; Mills, K.; Hole, K.H.; Inderberg, E.M. Phase I/IIa Clinical Trial of a Novel hTERT Peptide Vaccine in Men with Metastatic Hormone-Naive Prostate Cancer. Cancer Immunol. Immunother. 2017, 66, 891–901. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakken, S.; Lilleby, W.; Switlyk, M.D.; Knudsen, K.E.; Lilleby, O.; Zhao, S.; Kaveh, F.; Ekstrøm, P.O.; Urbanucci, A.; Hovig, E. The Quandary of DNA-Based Treatment Assessment in De Novo Metastatic Prostate Cancer in the Era of Precision Oncology. J. Pers. Med. 2021, 11, 330. https://doi.org/10.3390/jpm11050330
Nakken S, Lilleby W, Switlyk MD, Knudsen KE, Lilleby O, Zhao S, Kaveh F, Ekstrøm PO, Urbanucci A, Hovig E. The Quandary of DNA-Based Treatment Assessment in De Novo Metastatic Prostate Cancer in the Era of Precision Oncology. Journal of Personalized Medicine. 2021; 11(5):330. https://doi.org/10.3390/jpm11050330
Chicago/Turabian StyleNakken, Sigve, Wolfgang Lilleby, Marta D. Switlyk, Karen E. Knudsen, Oscar Lilleby, Sen Zhao, Fatemeh Kaveh, Per O. Ekstrøm, Alfonso Urbanucci, and Eivind Hovig. 2021. "The Quandary of DNA-Based Treatment Assessment in De Novo Metastatic Prostate Cancer in the Era of Precision Oncology" Journal of Personalized Medicine 11, no. 5: 330. https://doi.org/10.3390/jpm11050330
APA StyleNakken, S., Lilleby, W., Switlyk, M. D., Knudsen, K. E., Lilleby, O., Zhao, S., Kaveh, F., Ekstrøm, P. O., Urbanucci, A., & Hovig, E. (2021). The Quandary of DNA-Based Treatment Assessment in De Novo Metastatic Prostate Cancer in the Era of Precision Oncology. Journal of Personalized Medicine, 11(5), 330. https://doi.org/10.3390/jpm11050330