
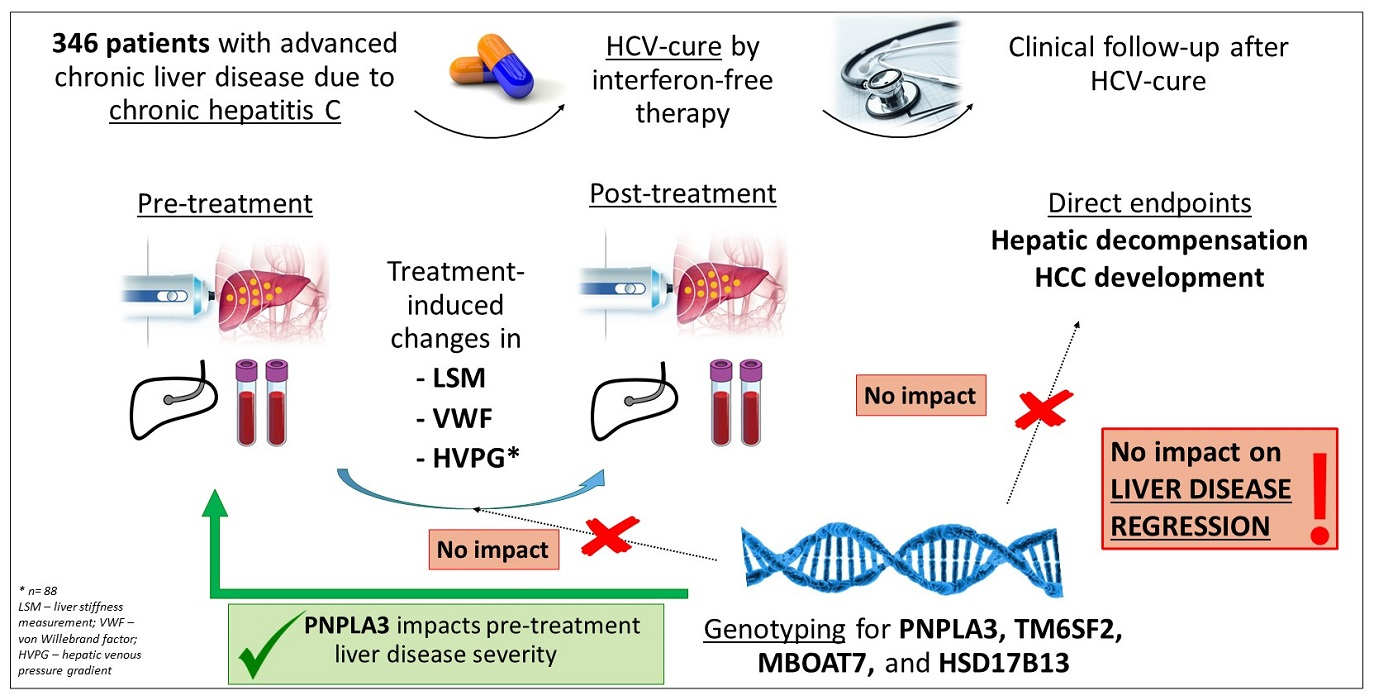
Influence of Genetic Variants on Disease Regression and Outcomes in HCV-Related Advanced Chronic Liver Disease after SVR

 , , , , , , , , , and
, , , , , , , , , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Study Population and Design
2.2. Genotyping and Assessment of Linkage Disequilibrium
2.3. HVPG Measurement (Cohort A)
2.4. Clinical and Laboratory Parameters (Cohorts A and B)
2.5. Clinical Events during FU (Cohort B)
2.6. HCV Therapy, Statistical Analyses and Ethics
3. Results
3.1. Patient Characteristics
3.2. Genetic Variants and Changes in HVPG (Cohort A)
3.3. Genetic Variants and Changes in Non-Invasive Surrogates of Portal Hypertension (Cohort B)
3.4. Hepatic Decompensation during Follow-Up (Cohort B)
3.5. De-Novo Hepatocellular Carcinoma Risk as Well as Transplant-Free and Liver-Related Mortality
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mandorfer, M.; Schwabl, P.; Steiner, S.; Scheiner, B.; Chromy, D.; Bucsics, T.; Stättermayer, A.F.; Aichelburg, M.C.; Grabmeier-Pfistershammer, K.; Trauner, M.; et al. Interferon-free treatment with sofosbuvir/daclatasvir achieves sustained virologic response in 100% of HIV/hepatitis C virus-coinfected patients with advanced liver disease. AIDS 2016, 30, 1039–1047. [Google Scholar] [CrossRef]
- Mandorfer, M.; Schwabl, P.; Steiner, S.; Reiberger, T.; Peck-Radosavljevic, M. Advances in the management of HIV/HCV coinfection. Hepatol. Int. 2016, 10, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Mandorfer, M.; Kozbial, K.; Freissmuth, C.; Schwabl, P.; Stättermayer, A.F.; Reiberger, T.; Beinhardt, S.; Schwarzer, R.; Trauner, M.; Ferlitsch, A.; et al. Interferon-free regimens for chronic hepatitis C overcome the effects of portal hypertension on virological responses. Aliment. Pharmacol. Ther. 2015, 42, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Kozbial, K.; Mandorfer, M.; Hofer, H. HCV targeting of patients with cirrhosis. J. Hepatol. 2015, 63, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- George, S.L.; Bacon, B.R.; Brunt, E.M.; Mihindukulasuriya, K.L.; Hoffmann, J.; Di Bisceglie, A.M. Clinical, virologic, histologic, and biochemical outcomes after successful HCV therapy: A 5-year follow-up of 150 patients. Hepatology 2008, 49, 729–738. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Aghemo, A.; Rumi, M.G.; Ronchi, G.; Donato, M.F.; Paradis, V.; Colombo, M.; Bedossa, P. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology 2012, 56, 532–543. [Google Scholar] [CrossRef]
- Mandorfer, M.; Kozbial, K.; Schwabl, P.; Freissmuth, C.; Schwarzer, R.; Stern, R.; Chromy, D.; Stättermayer, A.F.; Reiberger, T.; Beinhardt, S.; et al. Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J. Hepatol. 2016, 65, 692–699. [Google Scholar] [CrossRef]
- Mandorfer, M.; Kozbial, K.; Schwabl, P.; Chromy, D.; Semmler, G.; Stättermayer, A.F.; Pinter, M.; Hernández-Gea, V.; Fritzer-Szekeres, M.; Steindl-Munda, P.; et al. Changes in Hepatic Venous Pressure Gradient Predict Hepatic Decompensation in Patients Who Achieved Sustained Virologic Response to Interferon-Free Therapy. Hepatology 2020, 71, 1023–1036. [Google Scholar] [CrossRef]
- Lens, S.; Alvarado-Tapias, E.; Mariño, Z.; Londoño, M.-C.; Llop, E.; Martinez, J.; Fortea, J.I.; Ibañez, L.; Ariza, X.; Baiges, A.; et al. Effects of All-Oral Anti-Viral Therapy on HVPG and Systemic Hemodynamics in Patients With Hepatitis C Virus-Associated Cirrhosis. Gastroenterology 2017, 153, 1273–1283.e1. [Google Scholar] [CrossRef]
- Lens, S.; Baiges, A.; Alvarado-Tapias, E.; Llop, E.; Martinez, J.; Fortea, J.I.; Ibáñez-Samaniego, L.; Mariño, Z.; Rodríguez-Tajes, S.; Gallego, A.; et al. Clinical outcome and hemodynamic changes following HCV eradication with oral antiviral therapy in patients with clinically significant portal hypertension. J. Hepatol. 2020, 73, 1415–1424. [Google Scholar] [CrossRef]
- Bosch, J.; Groszmann, R.J.; Shah, V.H. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J. Hepatol. 2015, 62, S121–S130. [Google Scholar] [CrossRef] [PubMed]
- Mauro, E.; Crespo, G.; Montironi, C.; Londoño, M.-C.; Hernández-Gea, V.; Ruiz, P.; Sastre, L.; Lombardo, J.; Mariño, Z.; Díaz, A.; et al. Portal pressure and liver stiffness measurements in the prediction of fibrosis regression after sustained virological response in recurrent hepatitis C. Hepatology 2018, 67, 1683–1694. [Google Scholar] [CrossRef]
- Mandorfer, M.; Hernández-Gea, V.; García-Pagán, J.C.; Reiberger, T. Noninvasive Diagnostics for Portal Hypertension: A Comprehensive Review. Semin. Liver Dis. 2020, 40, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Semmler, G.; Binter, T.; Kozbial, K.; Schwabl, P.; Hametner-Schreil, S.; Zanetto, A.; Gavasso, S.; Chromy, D.; Bauer, D.J.M.; Simbrunner, B.; et al. Non-invasive risk stratification after HCV-eradication in patients with advanced chronic liver disease. Hepatology (Baltimore, Md.) 2020. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Trépo, E.; Pradat, P.; Potthoff, A.; Momozawa, Y.; Quertinmont, E.; Gustot, T.; Lemmers, A.; Berthillon, P.; Amininejad, L.; Chevallier, M.; et al. Impact of patatin-like phospholipase-3 (rs738409 C>G) polymorphism on fibrosis progression and steatosis in chronic hepatitis C. Hepatology 2011, 54, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Mandorfer, M.; Payer, B.A.; Schwabl, P.; Steiner, S.; Ferlitsch, A.; Aichelburg, M.C.; Stättermayer, A.F.; Ferenci, P.; Obermayer-Pietsch, B.; Grabmeier-Pfistershammer, K.; et al. Revisiting liver disease progression in HIV/HCV-coinfected patients: The influence of vitamin D, insulin resistance, immune status, IL28B and PNPLA3. Liver Int. 2014, 35, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Rumi, M.; Galmozzi, E.; Aghemo, A.; Del Menico, B.; De Nicola, S.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Rametta, R.; et al. Patatin-Like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology 2010, 53, 791–799. [Google Scholar] [CrossRef]
- Rüeger, S.; Bochud, P.-Y.; Dufour, J.-F.; Müllhaupt, B.; Semela, D.; Heim, M.H.; Moradpour, D.; Cerny, A.; Malinverni, R.; Booth, D.R.; et al. Impact of common risk factors of fibrosis progression in chronic hepatitis C. Gut 2015, 64, 1605–1615. [Google Scholar] [CrossRef]
- Mandorfer, M.; Scheiner, B.; Stättermayer, A.F.; Schwabl, P.; Paternostro, R.; Bauer, D.; Schaefer, B.; Zoller, H.; Peck-Radosavljevic, M.; Trauner, M.; et al. Impact of patatin-like phospholipase domain containing 3 rs738409 G/G genotype on hepatic decompensation and mortality in patients with portal hypertension. Aliment. Pharmacol. Ther. 2018, 48, 451–459. [Google Scholar] [CrossRef]
- Milano, M.; Aghemo, A.; Mancina, R.M.; Fischer, J.; Dongiovanni, P.; De Nicola, S.; Fracanzani, A.L.; D’Ambrosio, R.; Maggioni, M.; De Francesco, R.; et al. Transmembrane 6 superfamily member 2 gene E167K variant impacts on steatosis and liver damage in chronic hepatitis C patients. Hepatology 2015, 62, 111–117. [Google Scholar] [CrossRef]
- Thabet, K.; Asimakopoulos, A.; Shojaei, M.; Romero-Gomez, M.; Mangia, A.; Irving, W.L.; Berg, T.; Dore, G.J.; Grønbæk, H.; Sheridan, D.; et al. MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat. Commun. 2016, 7, 12757. [Google Scholar] [CrossRef]
- About, F.; Abel, L.; Cobat, A. HCV-Associated Liver Fibrosis and HSD17B13. New Engl. J. Med. 2018, 379, 1875–1876. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, B.; Stättermayer, A.F.; Schwabl, P.; Bucsics, T.; Paternostro, R.; Bauer, D.; Simbrunner, B.; Schmidt, R.; Marculescu, R.; Ferlitsch, A.; et al. Impact of HSD17B13 rs72613567 genotype on hepatic decompensation and mortality in patients with portal hypertension. Liver Int. 2020, 40, 393–404. [Google Scholar] [CrossRef] [PubMed]
- de Franchis, R. Expanding consensus in portal hypertension. J. Hepatol. 2015, 63, 743–752. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Rautou, P.-E.; Schouten, J.; Rubbia-Brandt, L.; Leebeek, F.; Trebicka, J.; Murad, S.D.; Vilgrain, V.; Hernandez-Gea, V.; Nery, F.; et al. Porto-sinusoidal vascular disease: Proposal and description of a novel entity. Lancet Gastroenterol. Hepatol. 2019, 4, 399–411. [Google Scholar] [CrossRef]
- Reiberger, T.; Schwabl, P.; Trauner, M.; Peck-Radosavljevic, M.; Mandorfer, M. Measurement of the Hepatic Venous Pressure Gradient and Transjugular Liver Biopsy. J. Vis. Exp. 2020. [Google Scholar] [CrossRef]
- Reiberger, T.; Püspök, A.; Schoder, M.; Baumann-Durchschein, F.; Bucsics, T.; Datz, C.; Dolak, W.; Ferlitsch, A.; Finkenstedt, A.; Graziadei, I.; et al. Austrian consensus guidelines on the management and treatment of portal hypertension (Billroth III). Wien. Klin. Wochenschr. 2017, 129, 135–158. [Google Scholar] [CrossRef]
- Schwabl, P.; Bota, S.; Salzl, P.; Mandorfer, M.; Payer, B.A.; Ferlitsch, A.; Stift, J.; Wrba, F.; Trauner, M.; Peck-Radosavljevic, M.; et al. New reliability criteria for transient elastography increase the number of accurate measurements for screening of cirrhosis and portal hypertension. Liver Int. 2014, 35, 381–390. [Google Scholar] [CrossRef]
- Mandorfer, M.; Reiberger, T.; Peck-Radosavljevic, M. Monitoring the Evolution of Portal Hypertension After Sustained Virologic Response. Gastroenterology 2018, 154, 1550–1551. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Mandorfer, M.; Schwabl, P.; Paternostro, R.; Pomej, K.; Bauer, D.; Thaler, J.; Ay, C.; Quehenberger, P.; Fritzer-Szekeres, M.; Peck-Radosavljevic, M.; et al. Von Willebrand factor indicates bacterial translocation, inflammation, and procoagulant imbalance and predicts complications independently of portal hypertension severity. Aliment. Pharmacol. Ther. 2018, 47, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Piecha, F.; Mandorfer, M.; Peccerella, T.; Ozga, A.-K.; Poth, T.; Vonbank, A.; Seitz, H.K.; Rausch, V.; Reiberger, T.; Mueller, S. Pharmacological decrease of liver stiffness is pressure-related and predicts long-term clinical outcome. Am. J. Physiol. Liver Physiol. 2018, 315, G484–G494. [Google Scholar] [CrossRef]
- Corma-Gómez, A.; Macías, J.; Téllez, F.; Freyre-Carrillo, C.; Morano, L.; Rivero-Juárez, A.; Ríos, M.J.; Alados, J.C.; Vera-Méndez, F.J.; Merchante, N.; et al. Liver stiffness at the time of sustained virological response predicts the clinical outcome in HIV/HCV-coinfected patients with advanced fibrosis treated with direct-acting antivirals. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Schwarzer, R.; Reiberger, T.; Mandorfer, M.; Kivaranovic, D.; Hametner, S.; Hametner, S.; Paternostro, R.; Scheiner, B.; Schneeweiss-Friedl, J.; Trauner, M.; et al. The von Willebrand Factor antigen to platelet ratio (VITRO) score predicts hepatic decompensation and mortality in cirrhosis. J. Gastroenterol. 2019, 55, 533–542. [Google Scholar] [CrossRef]
- Dunn, W.; Vittal, A.; Zhao, J.; He, J.; Chakraborty, S.; Whitener, M.; Fohn, S.; Ash, R.; Taylor, R.M.; Olyaee, M.; et al. PNPLA3gene predicts clinical recovery after sustained virological response in decompensated hepatitis C cirrhosis. BMJ Open Gastroenterol. 2019, 6, e000241. [Google Scholar] [CrossRef]
- Krassenburg, L.A.; Maan, R.; Ramji, A.; Manns, M.P.; Cornberg, M.; Wedemeyer, H.; de Knegt, R.J.; Hansen, B.E.; Janssen, H.L.; de Man, R.A.; et al. Clinical outcomes following DAA therapy in patients with HCV-related cirrhosis depend on disease severity. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Atkinson, S.R.; Way, M.J.; McQuillin, A.; Morgan, M.Y.; Thursz, M.R. Homozygosity for rs738409:G in PNPLA3 is associated with increased mortality following an episode of severe alcoholic hepatitis. J. Hepatol. 2017, 67, 120–127. [Google Scholar] [CrossRef]
- Huang, C.-F.; Dai, C.-Y.; Yeh, M.-L.; Huang, C.-I.; Tai, C.-M.; Hsieh, M.-H.; Liang, P.-C.; Lin, Y.-H.; Hsieh, M.-Y.; Yang, H.-L.; et al. Association of diabetes and PNPLA3 genetic variants with disease severity of patients with chronic hepatitis C virus infection. J. Hepatol. 2015, 62, 512–518. [Google Scholar] [CrossRef]
- Scheiner, B.; Mandorfer, M.; Schwabl, P.; Payer, B.A.; Bucsics, T.; Bota, S.; Aichelburg, M.C.; Grabmeier-Pfistershammer, K.; Stättermayer, A.; Ferenci, P.; et al. The Impact of PNPLA3 rs738409 SNP on Liver Fibrosis Progression, Portal Hypertension and Hepatic Steatosis in HIV/HCV Coinfection. PLoS ONE 2015, 10, e0143429. [Google Scholar] [CrossRef]
- Huang, Z.; Guo, X.; Zhang, G.; Liang, L.; Nong, B. Correlation between PNPLA3 rs738409 polymorphism and hepatocellular carcinoma: A meta-analysis of 10,330 subjects. Int. J. Biol. Markers 2019, 34, 117–122. [Google Scholar] [CrossRef]
- Pons, M.; Rodríguez-Tajes, S.; Esteban, J.I.; Mariño, Z.; Vargas, V.; Lens, S.; Buti, M.; Augustin, S.; Forns, X.; Mínguez, B.; et al. Non-invasive prediction of liver-related events in patients with HCV-associated compensated advanced chronic liver disease after oral antivirals. J. Hepatol. 2020, 72, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, E.; D’Ambrosio, R.; Iavarone, M.; Sangiovanni, A.; Aghemo, A.; Soffredini, R.; Borghi, M.; Lunghi, G.; Colombo, M.; Lampertico, P. Factors Associated With Increased Risk of De Novo or Recurrent Hepatocellular Carcinoma in Patients With Cirrhosis Treated With Direct-Acting Antivirals for HCV Infection. Clin. Gastroenterol. Hepatol. 2019, 17, 1183–1191.e7. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Beste, L.A.; Green, P.K.; Singal, A.G.; Tapper, E.B.; Waljee, A.K.; Sterling, R.K.; Feld, J.J.; Kaplan, D.E.; Taddei, T.H.; et al. Increased Risk for Hepatocellular Carcinoma Persists Up to 10 Years After HCV Eradication in Patients With Baseline Cirrhosis or High FIB-4 Scores. Gastroenterology 2019, 157, 1264–1278.e4. [Google Scholar] [CrossRef] [PubMed]
- Chromy, D.; Mandorfer, M.; Bucsics, T.; Schwabl, P.; Bauer, D.; Scheiner, B.; Schmidbauer, C.; Lang, G.F.; Szekeres, T.; Ferenci, P.; et al. Prevalence and Predictors of Hepatic Steatosis in Patients with HIV/HCV Coinfection and the Impact of HCV Eradication. AIDS Patient Care STDs 2019, 33, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.; O’Neil, M.; Zhao, J.; Wu, C.H.; Roberts, B.; Chakraborty, S.; Sherman, C.; Weaver, B.; Taylor, R.; Olson, J.; et al. Donor PNPLA3 rs738409 genotype affects fibrosis progression in liver transplantation for hepatitis C. Hepatology 2014, 59, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Castera, L.; Hézode, C.; Roudot-Thoraval, F.; Bastie, A.; Zafrani, E.-S.; Pawlotsky, J.-M.; Dhumeaux, D. Worsening of steatosis is an independent factor of fibrosis progression in untreated patients with chronic hepatitis C and paired liver biopsies. Gut 2003, 52, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Leandro, G.; Mangia, A.; Hui, J.; Fabris, P.; Rubbia–Brandt, L.; Colloredo, G.; Adinolfi, L.E.; Asselah, T.; Jonsson, J.R.; Smedile, A.; et al. Relationship Between Steatosis, Inflammation, and Fibrosis in Chronic Hepatitis C: A Meta-Analysis of Individual Patient Data. Gastroenterology 2006, 130, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Mandorfer, M.; Steiner, S.; Scheiner, B.; Chromy, D.; Herac, M.; Bucsics, T.; Hayden, H.; Grabmeier-Pfistershammer, K.; Ferlitsch, A.; et al. Interferon-free regimens improve portal hypertension and histological necroinflammation in HIV/HCV patients with advanced liver disease. Aliment. Pharmacol. Ther. 2016, 45, 139–149. [Google Scholar] [CrossRef]
- Semmler, G.; Stift, J.; Scheiner, B.; Wöran, K.; Schwabl, P.; Paternostro, R.; Bucsics, T.; Stättermayer, A.F.; Pinter, M.; Ferlitsch, A.; et al. Performance of Controlled Attenuation Parameter in Patients with Advanced Chronic Liver Disease and Portal Hypertension. Dig. Dis. Sci. 2019, 64, 3642–3651. [Google Scholar] [CrossRef] [PubMed]
- EASL recommendations on treatment of Hepatitis C 2020. J. Hepatol. 2020, 73, 1170–1218. [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | All Patients, n = 346 | C/C, n = 173 | G/C or G/G, n = 173 | p Value |
|---|---|---|---|---|
| Age, years | 55.4 ± 10.7 | 57.2 ± 10.8 | 53.7 ± 10.4 | 0.002 |
| Sex | ||||
| Male | 230 (66.5%) | 109 (63.0%) | 121 (69.9%) | 0.172 |
| Female | 116 (33.5%) | 64 (37.0%) | 52 (30.1%) | |
| History of hepatic decompensation | 44 (12.7%) | 18 (10.4%) | 26 (15.0%) | 0.197 |
| Varices | 92 (26.6%) | 36 (20.8%) | 56 (32.4%) | 0.015 |
| Small | 46 (13.5%) | 18 (10.4%) | 29 (16.8%) | 0.051 |
| Large | 43 (12.6%) | 18 (10.4%) | 27 (15.6%) | |
| BL-CTP score, points | 5 ± 1 | 5 ± 1 | 6 ± 1 | 0.015 |
| Stage A | 304 (87.9%) | 161 (93.1%) | 143 (82.7%) | 0.003 |
| Stage B/C | 42 (12.1%) | 12 (6.9%) | 30 (17.3%) | |
| BL-MELD score, points | 8.9 ± 2.8 | 8.4 ± 2.4 | 9.3 ± 3.1 | 0.001 |
| BL-LSM 1, kPa | 17.7 (11.8–28.5) | 16.9 (11.8–26.6) | 20.9 (11.8–32.8) | 0.098 |
| Evidence of CSPH 2 | 181 (52.3%) | 76 (43.9%) | 105 (60.7%) | 0.002 |
| BL-PLT, G × L−1 | 138 ± 65 | 145 ± 66 | 130 ± 63 | 0.045 |
| BL-VWF 3, % | 239 (173–321) | 232 (167–307) | 249 (185–334) | 0.127 |
| BL-VITRO 3 | 1.91 (1.10–3.38) | 1.65 (0.99–3.23) | 2.05 (1.18–3.87) | 0.033 |
| BL-bilirubin, mg × dL−1 | 0.73 (0.54–1.13) | 0.70 (0.55–0.98) | 0.81 (0.53–1.23) | 0.072 |
| BL-creatinine, mg × dL−1 | 0.78 (0.68–0.91) | 0.79 (0.70–0.92) | 0.77 (0.67–0.91) | 0.335 |
| BL-albumin, g × L−1 | 40.6 ± 4.9 | 40.7 ± 4.9 | 40.4 ± 5.0 | 0.585 |
| BL-INR | 1.18 ± 0.27 | 1.13 ± 0.18 | 1.22 ± 0.33 | 0.003 |
| BL-AST, U × L−1 | 67 (43–101) | 64 (41–97) | 71 (45–105) | 0.260 |
| BL-ALT, U × L−1 | 69 (42–107) | 64 (43–105) | 73 (38–109) | 0.528 |
| BL-GGT, U × L−1 | 93 (52–160) | 89 (50–165) | 94 (56–149) | 0.800 |
| Patient Characteristics | C/C, n = 173 | G/C or G/G, n = 173 | p Value * | p Value ** |
|---|---|---|---|---|
| BL-liver stiffness 1, kPa | 16.8 (11.8–26.6) | 20.9 (11.8–32.7) | 0.129 | - |
| Absolute Δ LSM 1, kPa | −3.4 (−7.4–(0.2)) | −4.4 (-9.0-(−0.5)) | 0.620 | 0.720 |
| Relative Δ LSM 1, % | −23.1 (−38.9–(−1.5)) | −20.4 (−45.3–(−0.3)) | 0.738 | 0.885 |
| FU-LSM1, kPa | 13.5 (8.8–19.8) | 14.2 (8.6–26.3) | 0.491 | 0.720 |
| BL-CAP2, dB × m−1 | 253 ± 60 | 242 ± 60 | 0.246 | - |
| BL prevalence of hepatic steatosis 2, % | 52 (57.8%) | 43 (47.3%) | 0.156 | - |
| Absolute Δ CAP2, dB × m−1 | −6 (64) | 4 (59) | 0.087 | 0.145 |
| Relative Δ CAP 2, % | −1.9 (25.1) | 1.5 (27.0) | 0.096 | 0.312 |
| FU-CAP2, dB × m−1 | 243 ± 52 | 246 ± 52 | 0.643 | 0.145 |
| FU prevalence of hepatic steatosis 2, % | 43 (47.8%) | 38 (41.8%) | 0.415 | - |
| BL-PLT, G × L−1 | 145 ± 66 | 130 ± 63 | 0.045 | - |
| Absolute Δ PLT, G × L−1 | 11 (−3–29) | 6 (−5–20) | 0.069 | 0.154 |
| Relative Δ PLT, % | 8.5 (−2.8–20.3) | 5.8 (−5.5–17.7) | 0.130 | 0.093 |
| FU-PLT, G × L−1 | 160 ± 72 | 143 ± 74 | 0.031 | 0.154 |
| BL-VWF 3, % | 231 (167–304) | 251 (185–339) | 0.065 | - |
| Absolute Δ VWF 3, % | −36 (−73–(-10)) | −39 (−91–(11)) | 0.624 | 0.622 |
| Relative Δ VWF 3, % | −18.3 (−30.3–(−5.1)) | −17.8 (−31.0–(−5.7)) | 0.927 | 0.908 |
| FU-VWF 3, % | 178 (135–241) | 193 (145–269) | 0.231 | 0.622 |
| BL-VITRO 3 | 1.59 (0.97–3.22) | 2.06 (1.19–4.00) | 0.015 | - |
| Absolute Δ VITRO 3 | −0.31 (−1.00–(−0.07)) | −0.35 (−0.94–(0–12)) | 0.639 | 0.498 |
| Relative Δ VITRO 3, % | −24.9 (−40.6–(−6.1)) | −21.7 (−37.5–(6.5)) | 0.323 | 0.480 |
| FU-VITRO 3 | 1.12 (0.67–2.17) | 1.64 (0.79–2.71) | 0.012 | 0.498 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semmler, G.; Binter, T.; Kozbial, K.; Schwabl, P.; Chromy, D.; Bauer, D.; Simbrunner, B.; Müllner-Bucsics, T.; Scheiner, B.; Stättermayer, A.; et al. Influence of Genetic Variants on Disease Regression and Outcomes in HCV-Related Advanced Chronic Liver Disease after SVR. J. Pers. Med. 2021, 11, 281. https://doi.org/10.3390/jpm11040281
Semmler G, Binter T, Kozbial K, Schwabl P, Chromy D, Bauer D, Simbrunner B, Müllner-Bucsics T, Scheiner B, Stättermayer A, et al. Influence of Genetic Variants on Disease Regression and Outcomes in HCV-Related Advanced Chronic Liver Disease after SVR. Journal of Personalized Medicine. 2021; 11(4):281. https://doi.org/10.3390/jpm11040281
Chicago/Turabian StyleSemmler, Georg, Teresa Binter, Karin Kozbial, Philipp Schwabl, David Chromy, David Bauer, Benedikt Simbrunner, Theresa Müllner-Bucsics, Bernhard Scheiner, Albert Stättermayer, and et al. 2021. "Influence of Genetic Variants on Disease Regression and Outcomes in HCV-Related Advanced Chronic Liver Disease after SVR" Journal of Personalized Medicine 11, no. 4: 281. https://doi.org/10.3390/jpm11040281
APA StyleSemmler, G., Binter, T., Kozbial, K., Schwabl, P., Chromy, D., Bauer, D., Simbrunner, B., Müllner-Bucsics, T., Scheiner, B., Stättermayer, A., Pinter, M., Steindl-Munda, P., Trauner, M., Ferenci, P., Reiberger, T., & Mandorfer, M. (2021). Influence of Genetic Variants on Disease Regression and Outcomes in HCV-Related Advanced Chronic Liver Disease after SVR. Journal of Personalized Medicine, 11(4), 281. https://doi.org/10.3390/jpm11040281