
Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors?


{kind=link}
Abstract
1. Introduction
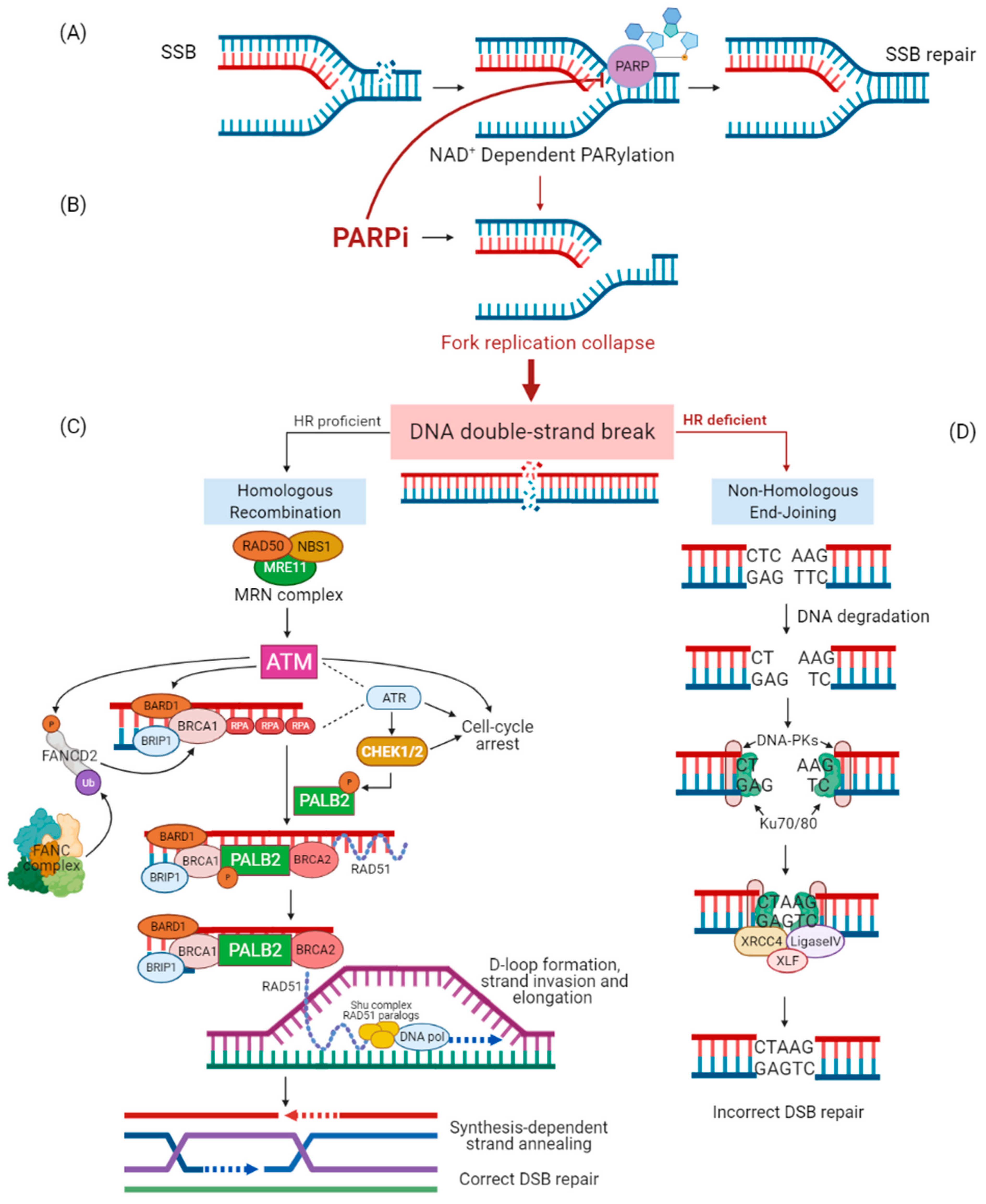
1.1. DNA Repair Mechanisms
1.2. PARP Inhibitor Treatments
1.3. PARPi Targeting HRR Genes other Than BRCA
2. The Different Role of High- and Moderate-Penetrance Genes
3. Results
3.1. How to Improve the Access to Genetic Counseling and Testing
3.2. How to Improve PARPi Response in gHRR Mutation Carriers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.C.; Côté, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240. [Google Scholar] [CrossRef]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Cell Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Desmond, A.; Kurian, A.W.; Gabree, M.; Mills, M.A.; Anderson, M.J.; Kobayashi, Y.; Horick, N.; Yang, S.; Shannon, K.M.; Tung, N.; et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol. 2015, 1, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, N.S.; Curcio, L.D.; Blakemore, C.A.; Bremner, A.K.; McFarland, R.E.; West, J.G.; Banks, K.C. Multigene panel testing detects equal rates of pathogenic BRCA1/2 mutations and has a higher diagnostic yield compared to limited BRCA1/2 analysis alone in patients at risk for hereditary breast cancer. Ann. Surg. Oncol. 2015, 22, 3282–3288. [Google Scholar] [CrossRef] [PubMed]
- Marabelli, M.; Cheng, S.C.; Parmigiani, G. Penetrance of ATM Gene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet. Epidemiol. 2016, 40, 425–431. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojć, B.; Deebniak, T.; Górski, B.; Blecharz, P.; et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef]
- Cybulski, C.; Górski, B.; Huzarski, T.; Masojć, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009, 324, 217. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Gruber, J.J.; Afghahi, A.; Hatton, A.; Scott, D.; McMillan, A.; Ford, J.M.; Telli, M.L. Talazoparib beyond BRCA: A phase II trial of talazoparib monotherapy in BRCA1 and BRCA2 wild-type patients with advanced HER2-negative breast cancer or other solid tumors with a mutation in homologous recombination (HR) pathway genes. J. Clin. Oncol. 2019, 37, 3006. [Google Scholar] [CrossRef]
- Yadav, S.; Kasi, P.M.; Bamlet, W.R.; Ho, T.P.; Polley, E.C.; Hu, C.; Hart, S.N.; Rabe, K.G.; Boddicker, N.J.; Gnanaolivu, R.D.; et al. Effect of Germline Mutations in Homologous Recombination Repair Genes on Overall Survival of Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2020, 26, 6505–6512. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar] [CrossRef]
- Buisson, R.; Dion-Côté, A.M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Dray, E.; Etchin, J.; Wiese, C.; Saro, D.; Williams, G.J.; Yu, X.; Galkin, V.E.; Liu, D.; Tsai, M.; Sy, S.M.H.; et al. Enhancement of the RAD51 Recombinase Activity by the Tumor Suppressor PALB2. Nat. Struct. Mol. Biol. 2011, 17, 1255–1259. [Google Scholar] [CrossRef]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef]
- Nepomuceno, T.C.; De Gregoriis, G.; de Oliveira, F.M.B.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. The Role of PALB2 in the DNA Damage Response and Cancer Predisposition. Int. J. Mol. Sci. 2017, 18, 1886. [Google Scholar] [CrossRef]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, W.; Sy, S.M.H.; Huen, M.S.Y. ATM-dependent phosphorylation of the Fanconi anemia protein PALB2 promotes the DNA damage response. J. Biol. Chem. 2015, 290, 27545–27556. [Google Scholar] [CrossRef]
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanović, M. The ATM signaling network in development and disease. Front. Genet. 2013, 4, 37. [Google Scholar] [CrossRef]
- Guleria, A.; Chandna, S. ATM kinase: Much more than a DNA damage responsive protein. DNA Repair 2016, 39, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM–CHK2 and ATR–CHK1 pathways for anticancer therapy. Mol. Cell Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, P.S.; Nielsen, S.; Olopade, O.I. USPSTF recommendations for BRCA1 and BRCA2 testing in the context of a transformative national cancer control plan. JAMA Netw. Open 2019, 2, e1910142. [Google Scholar] [CrossRef]
- Hull, L.E.; Haas, J.S.; Simon, S.R. Provider discussions of genetic tests with U.S. women at risk for a BRCA mutation. Am. J. Prev. Med. 2018, 54, 221–228. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic (Version 2–20 November 2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 24 February 2021).
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E.; ESMO Guidelines Committee. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27, v103–v110. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Bulsara, C.; Cohen, P.A.; Gryta, M.; Nichols, C.B.; Schofield, L.; O’Sullivan, S.; Pachter, N.; Hardcastle, S.J. Investigating barriers to genetic counseling and germline mutation testing in women with suspected hereditary breast and ovarian cancer syndrome and Lynch syndrome. Patient Educ. Couns. 2018, 101, 938–944. [Google Scholar] [CrossRef]
- PDQ Cancer Genetics Editorial Board. Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version. 4 December 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK65817/ (accessed on 24 February 2021).
- Vogel, R.I.; Niendorf, K.; Lee, H.; Petzel, S.; Lee, H.Y.; Geller, M.A. A qualitative study of barriers to genetic counseling and potential for mobile technology education among women with ovarian cancer. Hered. Cancer Clin. Pract. 2018, 16, 13. [Google Scholar] [CrossRef]
- Hann, K.E.J.; Freeman, M.; Fraser, L.; Waller, J.; Sanderson, S.C.; Rahman, B.; Side, L.; Gessler, S.; Lanceley, A.; PROMISE Study Team. Awareness, knowledge, perceptions, and attitudes towards genetic testing for cancer risk among ethnic minority groups: A systematic review. BMC Public Health 2017, 17, 503. [Google Scholar] [CrossRef]
- Jones, T.; McCarthy, A.M.; Kim, Y.; Armstrong, K. Predictors of BRCA1/2 genetic testing among Black women with breast cancer: A population-based study. Cancer Med. 2017, 6, 1787–1798. [Google Scholar] [CrossRef]
- Godard, B.; Kääriäinen, H.; Kristoffersson, U.; Tranebjaerg, L.; Coviello, D.; Aymé, S. Provision of genetic services in Europe: Current practices and issues. Eur. J. Hum. Genet. 2003, 11, S13–S48. [Google Scholar] [CrossRef][Green Version]
- Scherr, C.L.; Bomboka, L.; Nelson, A.; Pal, T.; Vadaparampil, S.T. Tracking the dissemination of a culturally targeted brochure to promote awareness of hereditary breast and ovarian cancer among Black women. Patient Educ. Couns. 2017, 100, 805–811. [Google Scholar] [CrossRef]
- Cohen, S.A.; Bradbury, A.; Henderson, V.; Hoskins, K.; Bednar, E.; Arun, B.K. Genetic Counseling and Testing in a Community Setting: Quality, Access, and Efficiency. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e34–e44. [Google Scholar] [CrossRef]
- Katsanis, S.H.; Katsanis, N. Molecular genetic testing and the future of clinical genomics. Nat. Rev. Genet. 2013, 14, 415–426. [Google Scholar] [CrossRef]
- Di Resta, C.; Galbiati, S.; Carrera, P.; Ferrari, M. Next-generation sequencing approach for the diagnosis of human diseases: Open challenges and new opportunities. EJIFCC 2018, 29, 4–14. [Google Scholar]
- Oplustilova, L.; Wolanin, K.; Mistrik, M.; Korinkova, G.; Simkova, D.; Bouchal, J.; Lenobel, R.; Bartkova, J.; Lau, A.; O’Connor, M.J.; et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle 2012, 11, 3837–3850. [Google Scholar] [CrossRef]
- Michels, J.; Vitale, I.; Saparbaev, M.; Castedo, M.; Kroemer, G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene 2014, 33, 3894–3907. [Google Scholar] [CrossRef] [PubMed]
- Szántó, M.; Bai, P. The role of ADP-ribose metabolism in metabolic regulation, adipose tissue differentiation, and metabolism. Genes Dev. 2020, 34, 321–340. [Google Scholar] [CrossRef]
- Ke, Y.; Wang, C.; Zhang, J.; Zhong, X.; Wang, R.; Zeng, X.; Ba, X. The Role of PARPs in Inflammation-and Metabolic-Related Diseases: Molecular Mechanisms and Beyond. Cells 2019, 8, 1047. [Google Scholar] [CrossRef]
- Tutt, A.; Stephens, C.; Frewer, P.; Pierce, A.; Rhee, J.; So, K.; Ottesen, L.; Dean, E.; Hollingsworth, S.J. VIOLETTE: A randomized phase II study to assess DNA damage response inhibitors in combination with olaparib (Ola) vs Ola monotherapy in patients (pts) with metastatic, triple-negative breast cancer (TNBC) stratified by alterations in homologous recombination repair (HRR)-related genes. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Hamilton, E.; Falchook, G.S.; Wang, J.S.; Fu, S.; Oza, A.; Karen, S.; Imedio, E.R.; Kumar, S.; Ottesen, L.; Mugundu, G.M.; et al. Abstract CT025: Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: Dose escalation. Cancer Res. 2019, 79 (Suppl. 13), CT025. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortesi, L.; Piombino, C.; Toss, A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? J. Pers. Med. 2021, 11, 245. https://doi.org/10.3390/jpm11040245
Cortesi L, Piombino C, Toss A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? Journal of Personalized Medicine. 2021; 11(4):245. https://doi.org/10.3390/jpm11040245
Chicago/Turabian StyleCortesi, Laura, Claudia Piombino, and Angela Toss. 2021. "Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors?" Journal of Personalized Medicine 11, no. 4: 245. https://doi.org/10.3390/jpm11040245
APA StyleCortesi, L., Piombino, C., & Toss, A. (2021). Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? Journal of Personalized Medicine, 11(4), 245. https://doi.org/10.3390/jpm11040245