
Prognosis-Based Early Intervention Strategies to Resolve Exacerbation and Progressive Lung Function Decline in Cystic Fibrosis


Abstract
:1. Introduction
2. Sweat Chloride Test and Nasal Potential Difference Measurements in Cystic Fibrosis
3. Pulmonary Function Tests for Cystic Fibrosis Lung Disease Progression
4. Lung Clearance Index for Cystic Fibrosis Airway Disease Progression
5. Functional Lung Imaging Modalities for Evaluating Cystic Fibrosis Lung Disease Progression
6. Force and Impulse Oscillometry Measurements for Regional Lung Function Analysis
7. Electrical Impedance Tomography for Non-Invasive Regional Lung Function Analysis
8. Prognostic Biomarkers of Cystic Fibrosis Lung Disease
9. Prognosis-Based Intervention Strategies for Cystic Fibrosis Exacerbations and Lung Disease
10. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valle, C.W.; Vij, N. Can correcting the ΔF508-CFTR proteostasis-defect rescue CF lung disease? Curr. Mol. Med. 2012, 12, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Vij, N. The NF-kappaB signaling in cystic fibrosis lung disease: Pathophysiology and therapeutic potential. Discov. Med. 2010, 9, 346–356. [Google Scholar] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Mazur, S.; Zeitlin, P.L. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS ONE 2009, 4, e4664. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Fang, S.; Zeitlin, P.L. Selective inhibition of endoplasmic reticulum-associated degradation rescues DeltaF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: Therapeutic implications. J. Biol. Chem. 2006, 281, 17369–17378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cebotaru, L.; Vij, N.; Ciobanu, I.; Wright, J.; Flotte, T.; Guggino, W.B. Cystic fibrosis transmembrane regulator missing the first four transmembrane segments increases wild type and DeltaF508 processing. J. Biol. Chem. 2008, 283, 21926–21933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, I.; Vij, N. Therapeutic Strategies to Correct Proteostasis-Imbalance in Chronic Obstructive Lung Diseases. Curr. Mol. Med. 2012, 12, 807–814. [Google Scholar] [CrossRef]
- Belcher, C.; Vij, N. Protein processing and inflammatory signaling in Cystic Fibrosis: Challenges and therapeutic strategies. Curr. Mol. Med. 2010, 10, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.J.; Soong, G.; Bryan, R.; Saba, S.; Prince, A. Activation of NF-kappaB in airway epithelial cells is dependent on CFTR trafficking and Cl- channel function. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L71–L78. [Google Scholar] [CrossRef]
- Ulrich, M.; Worlitzsch, D.; Viglio, S.; Siegmann, N.; Iadarola, P.; Shute, J.K.; Geiser, M.; Pier, G.B.; Friedel, G.; Barr, M.L.; et al. Alveolar inflammation in cystic fibrosis. J. Cyst. Fibros. 2010, 9, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B.; Grout, M.; Zaidi, T.S. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc. Natl. Acad. Sci. USA 1997, 94, 12088–12093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, M.J.; Treharne, K.J.; Winter, A.K.; Cassidy, D.M.; Land, S.; Mehta, A. Expression of wild-type CFTR suppresses NF-kappaB-driven inflammatory signalling. PLoS ONE 2010, 5, e11598. [Google Scholar] [CrossRef] [Green Version]
- Le Henaff, C.; Mansouri, R.; Modrowski, D.; Zarka, M.; Geoffroy, V.; Marty, C.; Tarantino, N.; Laplatine, E.; Marie, P.J. Increased NF-κB Activity and Decreased Wnt/β-Catenin Signaling Mediate Reduced Osteoblast Differentiation and Function in ΔF508 Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Mice. J. Biol. Chem. 2015, 290, 18009–18017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef]
- Del Porto, P.; Cifani, N.; Guarnieri, S.; Di, Domenico, E.G.; Mariggiò, M.A.; Spadaro, F.; Guglietta, S.; Anile, M.; Venuta, F.; Quattrucci, S.; et al. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS ONE 2011, 6, e19970. [Google Scholar]
- Nilsson, H.E.; Dragomir, A.; Lazorova, L.; Johannesson, M.; Roomans, G.M. CFTR and tight junctions in cultured bronchial epithelial cells. Exp. Mol. Pathol. 2010, 88, 118–127. [Google Scholar] [CrossRef]
- Ni, I.; Ji, C.; Vij, N. Second-Hand Cigarette Smoke Impairs Bacterial Phagocytosis in Macrophages by Modulating CFTR Dependent Lipid-Rafts. PLoS ONE 2015, 10, e0121200. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Min, T.; Vij, N. Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L811–L820. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Min, T.; Mazur, S.; Vij, N. Critical Modifier Role of Membrane-Cystic Fibrosis Transmembrane Conductance Regulator-Dependent Ceramide Signaling in Lung Injury and Emphysema. J. Immunol. 2010, 186, 602–613. [Google Scholar] [CrossRef]
- Teichgräber, V.; Ulrich, M.; Endlich, N.; Riethmüller, J.; Wilker, B.; De Oliveira–Munding, C.C.; Van Heeckeren, A.M.; Barr, M.L.; Von Kürthy, G.; Schmid, K.W.; et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008, 14, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Muñoz, G.; Yu, M.A.; Lefrançais, E.; Mallavia, B.; Valet, C.; Tian, J.J.; Ranucci, S.; Wang, K.M.; Liu, Z.; Kwaan, N.; et al. Cystic fibrosis transmembrane conductance regulator dysfunction in platelets drives lung hyperinflammation. J. Clin. Investig. 2020, 130, 2041–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappenden, P.; Sadler, S.; Wildman, M. An Early Health Economic Analysis of the Potential Cost Effectiveness of an Adherence Intervention to Improve Outcomes for Patients with Cystic Fibrosis. PharmacoEconomics 2017, 35, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehote, G.; Vij, N. Autophagy Augmentation to Alleviate Immune Response Dysfunction, and Resolve Respiratory and COVID-19 Exacerbations. Cells 2020, 9, 1952. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; Rosenstein, B.J.; White, T.B.; Accurso, F.J.; Castellani, C.; Cutting, G.R.; Durie, P.R.; LeGrys, V.A.; Massie, J.; Parad, R.B.; et al. Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation Consensus Report. J. Pediatr. 2008, 153, S4–S14. [Google Scholar] [CrossRef] [Green Version]
- Soares, M.; Richardson, M.; Thorpe, J.; Owers-Bradley, J.; Siddiqui, S. Comparison of Forced and Impulse Oscillometry Measurements: A Clinical Population and Printed Airway Model Study. Sci. Rep. 2019, 9, 2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, M.; Joachim, C.; Wielpütz, M.O.; Mall, M.A. Comparison of lung clearance index determined by washout of N2 and SF6 in infants and preschool children with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 399–406. [Google Scholar] [CrossRef]
- Stahl, M.; Wielpütz, M.O.; Graeber, S.Y.; Joachim, C.; Sommerburg, O.; Kauczor, H.-U.; Puderbach, M.; Eichinger, M.; Mall, M.A. Comparison of Lung Clearance Index and Magnetic Resonance Imaging for Assessment of Lung Disease in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 349–359. [Google Scholar] [CrossRef]
- Parsons, D.; Donnelley, M. Will airway gene therapy for cystic fibrosis improve lung function? New imaging technologies can help us find out. Hum. Gene Ther. 2020, 31, 973–984. [Google Scholar] [CrossRef]
- Wielpütz, M.O.; Heußel, C.P.; Herth, F.J.; Kauczor, H.U. Radiological diagnosis in lung disease: Factoring treatment options into the choice of diagnostic modality. Dtsch. Arztebl. Int. 2014, 111, 181–187. [Google Scholar]
- Heidenreich, J.F.; Weng, A.M.; Metz, C.; Benkert, T.; Pfeuffer, J.; Hebestreit, H.; Bley, T.A.; Köstler, H.; Veldhoen, S. Three-dimensional Ultrashort Echo Time MRI for Functional Lung Imaging in Cystic Fibrosis. Radiology 2020, 296, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, P.M.; A De Jong, P.; Tiddens, H.A.W.M.; Lindblad, A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax 2008, 63, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, B.; Löhr, S.; Zhao, Z.; Falkenberg, C.; Ankermann, T.; Weiler, N.; Frerichs, I. Regional lung function testing in children using electrical impedance tomography. Pediatr. Pulmonol. 2018, 53, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Schullcke, B.; Gong, B.; Krueger-Ziolek, S.; Soleimani, M.; Mueller-Lisse, U.; Moeller, K. Structural-functional lung imaging using a combined CT-EIT and a Discrete Cosine Transformation reconstruction method. Sci. Rep. 2016, 6, 25951. [Google Scholar] [CrossRef]
- Jang, G.Y.; Ayoub, G.; Kim, Y.E.; Oh, T.I.; Chung, C.R.; Suh, G.Y.; Woo, E.J. Integrated EIT system for functional lung ventilation imaging. Biomed. Eng. Online 2019, 18, 83. [Google Scholar] [CrossRef] [Green Version]
- Inany, H.S.; Rettig, J.S.; Smallwood, C.D.; Arnold, J.H.; Walsh, B.K. Distribution of Ventilation Measured by Electrical Impedance Tomography in Critically Ill Children. Respir. Care 2020, 65, 590–595. [Google Scholar] [CrossRef]
- Breuer, O.; Caudri, D.; Stick, S.; Turkovic, L. Predicting disease progression in cystic fibrosis. Expert Rev. Respir. Med. 2018, 12, 905–917. [Google Scholar] [CrossRef]
- Vij, N.; Min, T.; Bodas, M.; Gorde, A.; Roy, I. Neutrophil targeted nano-drug delivery system for chronic obstructive lung diseases. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 2415–2427. [Google Scholar] [CrossRef]
- Vij, N.; Amoako, M.O.; Mazur, S.; Zeitlin, P.L. CHOP Transcription Factor Mediates IL-8 Signaling in Cystic Fibrosis Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR Expression in Human Neutrophils and the Phagolysosomal Chlorination Defect in Cystic Fibrosis†. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [Green Version]
- Hayes, E.; Pohl, K.; McElvaney, N.G.; Reeves, E.P. The Cystic Fibrosis Neutrophil: A Specialized Yet Potentially Defective Cell. Arch. Immunol. Ther. Exp. 2011, 59, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Parker, M.M.; Oster, R.A.; Bowler, R.P.; Dransfield, M.T.; Bhatt, S.P.; Cho, M.H.; Kim, V.; Curtis, J.L.; Martinez, F.J.; et al. Elevated circulating MMP-9 is linked to increased COPD exacerbation risk in SPIROMICS and COPDGene. JCI Insight 2018, 3, e123614. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.; O’Carroll, O.; Franciosi, A.N.; McElvaney, N.G. Factors Affecting Prognosis and Prediction of Outcome in Cystic Fibrosis Lung Disease.
- Vietri, L.; Fui, A.; Bergantini, L.; D’Alessandro, M.; Cameli, P.; Sestini, P.; Rottoli, P.; Bargagli, E. Serum amyloid A: A potential biomarker of lung disorders. Respir. Investig. 2020, 58, 21–27. [Google Scholar] [CrossRef]
- Ventura, J.C.; Hauschild, D.B.; Moreira, E.A.M.; Pereira, L.C.R.; Rosa, A.F.; Barbosa, E.; Ludwig-Neto, N.; Da Rosa, J.S.; Fröde, T.S.; Moreno, Y.M.F. C-reactive protein/albumin ratio is associated with lung function among children/adolescents with cystic fibrosis: A three-year longitudinal study. Sao Paulo Med. J. 2018, 136, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.C.; Kiley, J.P.; Noel, P.J.; Amur, S.; Burchard, E.G.; Clancy, J.P.; Galanter, J.M.; Inada, M.; Jones, T.K.; Kropski, J.A.; et al. Current Status and Future Opportunities in Lung Precision Medicine Research with a Focus on Biomarkers. An American Thoracic Society/National Heart, Lung, and Blood Institute Research Statement. Am. J. Respir. Crit. Care Med. 2018, 198, e116–e136. [Google Scholar] [CrossRef]
- Kerem, E.; Corey, M.; Kerem, B.S.; Rommens, J.; Markiewicz, D.; Levison, H.; Tsui, L.-C.; Durie, P. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508). N. Engl. J. Med. 1990, 323, 1517–1522. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.A.; De Rosa, G.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef] [Green Version]
- Schiffhauer, E.S.; Vij, N.; Kovbasnjuk, O.; Kang, P.W.; Walker, D.; Lee, S.; Zeitlin, P.L. Dual activation of CFTR and CLCN2 by lubiprostone in murine nasal epithelia. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L324–L331. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, K.D.; McKenzie, K.R.; Henderson, M.J.; Hawkins, C.E.; Vij, N.; Zeitlin, P.L. Lubiprostone activates non-CFTR-dependent respiratory epithelial chloride secretion in cystic fibrosis mice. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L933–L940. [Google Scholar] [CrossRef]
- Briggs, E.C.; Nguyen, T.; Wall, M.A.; Moran, A.R. Oral antimicrobial use in outpatient cystic fibrosis pulmonary exacerbation management: A single-center experience. Clin. Respir. J. 2012, 6, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wark, P.; Cookson, K.; Thiruchelvam, T.; Brannan, J.D.; Dorahy, D.J. Lumacaftor/ Ivacaftor improves exercise tolerance in patients with Cystic Fibrosis and severe airflow obstruction. BMC Pulm. Med. 2019, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, J.; Ruppel, G.; Shrestha, Y.; Kleinhenz, M. The diffusing capacity in adult cystic fibrosis. Respir. Med. 2003, 97, 606–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechtzin, N.; Mayer-Hamblett, N.; West, N.E.; Allgood, S.; Wilhelm, E.; Khan, U.; Aitken, M.L.; Ramsey, B.W.; Boyle, M.P.; Mogayzel, P.J., Jr.; et al. Home Monitoring of Patients with Cystic Fibrosis to Identify and Treat Acute Pulmonary Exacerbations. eICE Study Results. Am. J. Respir. Crit. Care Med. 2017, 196, 1144–1151. [Google Scholar] [CrossRef]
- Wielpütz, M.O.; Eichinger, M.; Biederer, J.; Wege, S.; Stahl, M.; Sommerburg, O.; Mall, M.A.; Kauczor, H.-U.; Puderbach, M. Imaging of Cystic Fibrosis Lung Disease and Clinical Interpretation. RöFo—Fortschr. Gebiet Röntgenstrahlen Bildgeb. Verfahr. 2016, 188, 834–845. [Google Scholar] [CrossRef] [Green Version]
- Hegi-Johnson, F.; Keall, P.; Barber, J.; Bui, C.; Kipritidis, J. Evaluating the accuracy of 4D-CT ventilation imaging: First comparison with Technegas SPECT ventilation. Med Phys. 2017, 44, 4045–4055. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Izumi, Y.; Watanabe, W.; Shimizu, Y.; Osada, H.; Honda, N.; Itoh, T.; Nakayama, M. Generation of ventilation/perfusion ratio map in surgical patients by dual-energy CT after xenon inhalation and intravenous contrast media. J. Cardiothorac. Surg. 2018, 13, 43. [Google Scholar] [CrossRef]
- Tanimura, K.; Hirai, T.; Sato, S.; Hasegawa, K.; Muro, S.; Kurosawa, H.; Mishima, M. Comparison of two devices for respiratory impedance measurement using a forced oscillation technique: Basic study using phantom models. J. Physiol. Sci. 2014, 64, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Meraz, E.; Nazeran, H.; Goldman, M.; Nava, P.; Diong, B. Impulse oscillometric features of lung function: Towards computer-aided classification of respiratory diseases in children. In Proceedings of the 2008 30th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Vancouver, BC, Canada, 21–24 August 2008; Volume 2008, pp. 2443–2446. [Google Scholar]
- Hamakawa, H.; Sakai, H.; Takahashi, A.; Bando, T.; Date, H. Multi-frequency Forced Oscillation Technique Using Impulse Oscillations: Can It Give Mechanical Information about the Lung Periphery? Neurotransm. Interact. Cogn. Funct. 2013, 765, 73–79. [Google Scholar] [CrossRef]
- Bednarek, M.; Grabicki, M.; Piorunek, T.; Batura-Gabryel, H. Current place of impulse oscillometry in the assessment of pulmonary diseases. Respir. Med. 2020, 170. [Google Scholar] [CrossRef]
- Bodas, M.; Mazur, S.; Min, T.; Vij, N. Inhibition of histone-deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respir. Res. 2018, 19, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodas, M.; Silverberg, D.; Walworth, K.; Brucia, K.; Vij, N. Augmentation of S-Nitrosoglutathione Controls Cigarette Smoke-Induced Inflammatory–Oxidative Stress and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis by Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function. Antioxid. Redox Signal. 2017, 27, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-T.; Yang, C.-M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 2012, 84, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Wagner, B.D.; Ziady, A.; Kelley, T.; Clancy, J.P.; Narvaez-Rivas, M.; Pilewski, J.; Joseloff, E.; Sha, W.; Zelnick, L.; et al. Utilizing centralized biorepository samples for biomarkers of cystic fibrosis lung disease severity. J. Cyst. Fibros. 2020, 19, 632–640. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Clancy, J.P.; Heltshe, S.L.; Ziady, A.; Kelley, T.; Accurso, F.; Pilewski, J.; Mayer-Hamblett, N.; Joseloff, E.; Sagel, S.D. Biomarkers for cystic fibrosis drug development. J. Cyst. Fibros. 2016, 15, 714–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, C.L.; Assani, K.D.; Rinehardt, H.; Albastroiu, F.; Zhang, S.; Shell, R.; Amer, A.O.; Schlesinger, L.S.; Kopp, B.T. Cysteamine-mediated clearance of antibiotic-resistant pathogens in human cystic fibrosis macrophages. PLoS ONE 2017, 12, e0186169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrier, C.; Rodger, C.; Robertson, J.; Kowalczuk, A.; Shand, N.; Fraser-Pitt, D.J.; Mercer, D.K.; O’Neil, D. Cysteamine (Lynovex®), a novel mucoactive antimicrobial & antibiofilm agent for the treatment of cystic fibrosis. Orphanet J. Rare Dis. 2014, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Pehote, G.; Bodas, M.; Brucia, K.; Vij, N. Cigarette Smoke Exposure Inhibits Bacterial Killing via TFEB-Mediated Autophagy Impairment and Resulting Phagocytosis Defect. Mediat. Inflamm. 2017, 2017, 3028082. [Google Scholar] [CrossRef]
- Skov, M.; Hansen, C.R.; Pressler, T. Cystic fibrosis—an example of personalized and precision medicine. APMIS 2019, 127, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Roy, I.; Vij, N. Nanodelivery in airway diseases: Challenges and therapeutic applications. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Personalized or Precision Medicine? The Example of Cystic Fibrosis. Front. Pharmacol. 2017, 8, 390. [Google Scholar] [CrossRef] [PubMed]
- Hagemeijer, M.C.; Siegwart, D.J.; Strug, L.J.; Cebotaru, L.; Torres, M.J.; Sofoluwe, A.; Beekman, J.M. Translational research to enable personalized treatment of cystic fibrosis. J. Cyst. Fibros. 2018, 17, S46–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foundation, C.F. Cystic Fibrosis Personalized Medicine Cystic Fibrosis Foundation. Available online: https://www.cff.org/What-is-CF/Genetics/Personalized-Medicine (accessed on 1 January 2021).
- Feng, L.B.; Grosse, S.D.; Green, R.F.; Fink, A.K.; Sawicki, G.S. Precision Medicine In Action: The Impact of Ivacaftor on Cystic Fibrosis–Related Hospitalizations. Health Aff. 2018, 37, 773–779. [Google Scholar] [CrossRef] [PubMed]
- de la Rosa, C.D.; Navarro, R.A.; Girón, M.R.M.; Montull, V.B.; Olveira, F.C.; Padilla, G.A.; Prados, S.C.; Quintana, G.E.; Sibila, V.O.; Celorrio, J.N.; et al. Cost of Hospitalizations due to Exacerbation in Patients with Non-Cystic Fibrosis Bronchiectasis. Respiration 2018, 96, 406–416. [Google Scholar]
- Rossi, G.A.; Morelli, P.; Galietta, L.J.; Colin, A.A. Airway microenvironment alterations and pathogen growth in cystic fibrosis. Pediatr. Pulmonol. 2019, 54, 497–506. [Google Scholar] [CrossRef]
- Vij, N. Nano-based theranostics for chronic obstructive lung diseases: Challenges and therapeutic potential. Expert Opin. Drug Deliv. 2011, 8, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Vij, N. Synthesis and Evaluation of Airway-Targeted PLGA-PEG Nanoparticles for Drug Delivery in Obstructive Lung Diseases. Toxic. Assess. 2020, 2118, 147–154. [Google Scholar] [CrossRef]
- Vij, N. Nano-based rescue of dysfunctional autophagy in chronic obstructive lung diseases. Expert Opin. Drug Deliv. 2017, 14, 483–489. [Google Scholar] [CrossRef]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Hayes, N.; Kopp, B.T.; Hill, C.L.; Lallier, S.W.; Schwartz, C.M.; Tadesse, M.; Alsudayri, A.; Reynolds, S.D. Cell Therapy for Cystic Fibrosis Lung Disease: Regenerative Basal Cell Amplification. Stem Cells Transl. Med. 2019, 8, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.E.; Miller, S.M.; Mascenik, T.M.; Lewis, C.A.; Dang, H.; Boggs, Z.H.; Tarran, R.; Randell, S.H. Assessing Human Airway Epithelial Progenitor Cells for Cystic Fibrosis Cell Therapy. Am. J. Respir. Cell Mol. Biol. 2020, 63, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.D.; Erle, D.J. Steps toward Cell Therapy for Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Duchesneau, P.; Waddell, T.K.; Karoubi, G. Cell-Based Therapeutic Approaches for Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 5219. [Google Scholar] [CrossRef] [PubMed]
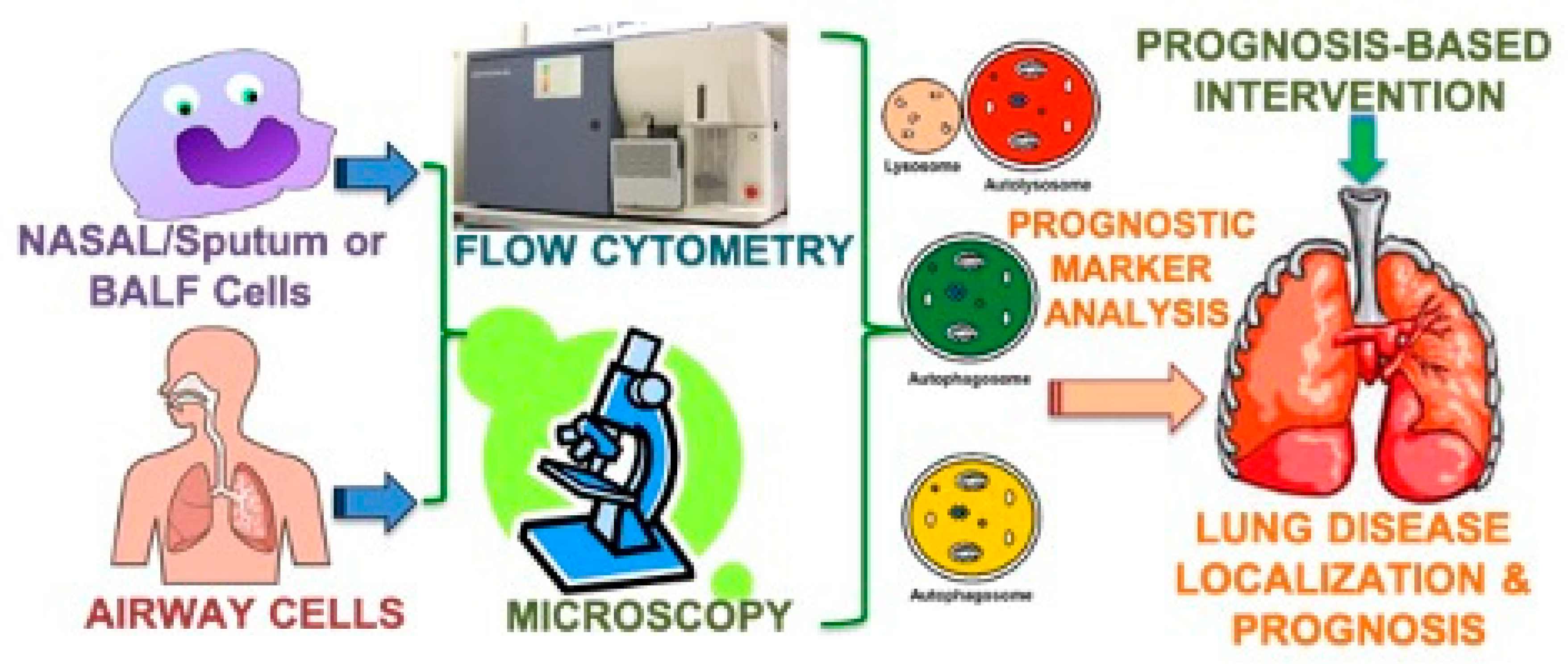

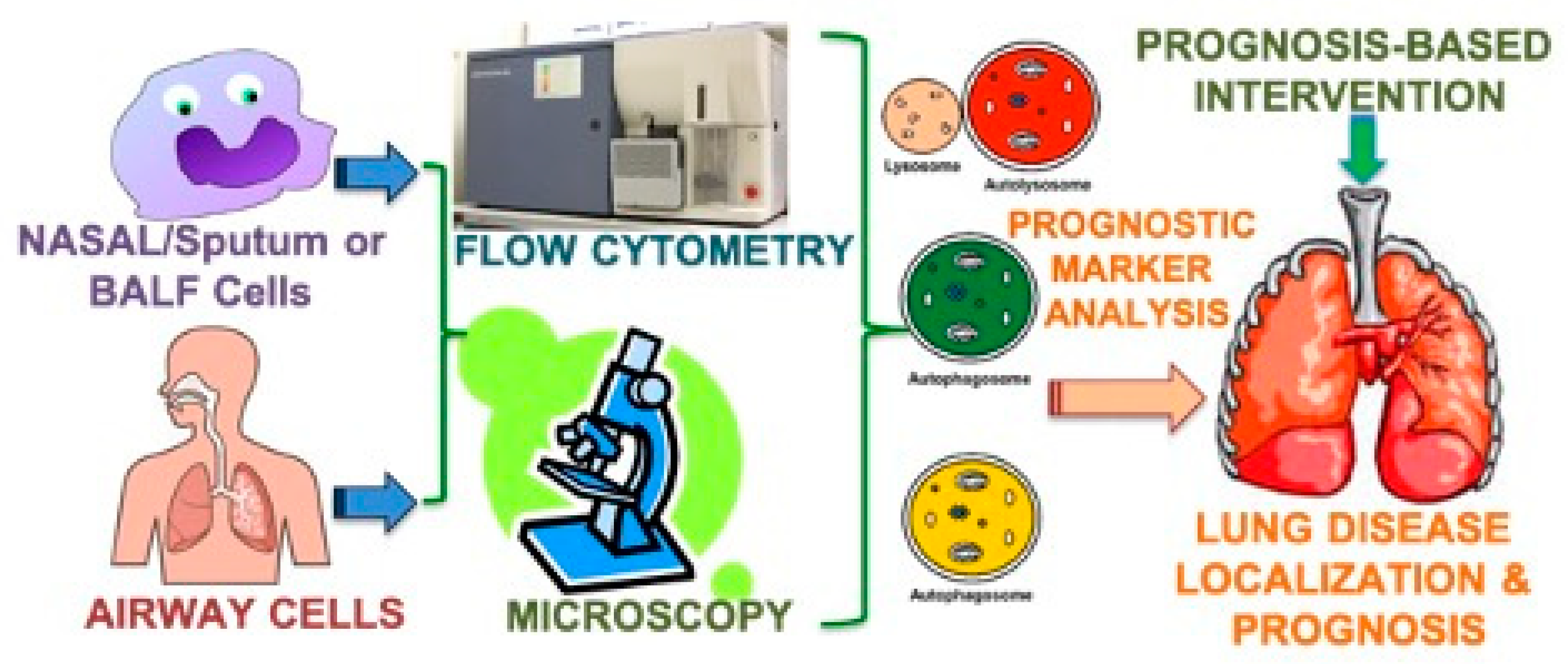{kind=link}
| Cystic Fibrosis & Lung Diagnostics | Lung Function/ Disease Analysis | Compatibility with Intervention | Predicts Initiation of Exacerbation | Setting | Associated Risks |
|---|---|---|---|---|---|
| Lung Imaging | Yes | Average | No | Radiology | High |
| PFT/Spirometry | Yes | Low | No | PFT Lab | No |
| LCI | Yes | Average | No | Bedside | No |
| Sweat Chloride | No | Low | No | Lab/Clinic | No |
| POC ID | No | High for ID | Yes | POC | No |
| FOT/IOS/EIT | Yes | High | No | Bedside | Minimal |
| Prognostics | Yes | Excellent | Yes | POC/Lab | No |
| Lung Diagnostic Advantage for CF | Cost ofDiagnostic/Test | Resolution/ Specificity | Level of Accuracy | Patient Compliance | Time to Complete |
|---|---|---|---|---|---|
| Thoracic X-Ray | Low | Low | Low | Not Required | 5 min |
| Thoracic PET/MRI | Very High | Moderate | Moderate | Required | 60–120 min |
| Thoracic CT Scans | High | High | High | Required | 20 min |
| PFT/Spirometry | Moderate | Low | Moderate | Patient Dependent | 30–60 min |
| POC ID | Moderate | High | High | Not Required | 15 min-72 h |
| LCI | Moderate | High | High | Required | 15–40 min |
| Sweat Chloride | Low | Average | Average | Not Required | 24–48 h |
| FOT/IOS/EIT | Moderate | High | High | Not Required | Real Time/Continuous |
| Prognostics | Moderate | Very High | High | Not Required | 15–40 min |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vij, N. Prognosis-Based Early Intervention Strategies to Resolve Exacerbation and Progressive Lung Function Decline in Cystic Fibrosis. J. Pers. Med. 2021, 11, 96. https://doi.org/10.3390/jpm11020096
Vij N. Prognosis-Based Early Intervention Strategies to Resolve Exacerbation and Progressive Lung Function Decline in Cystic Fibrosis. Journal of Personalized Medicine. 2021; 11(2):96. https://doi.org/10.3390/jpm11020096
Chicago/Turabian StyleVij, Neeraj. 2021. "Prognosis-Based Early Intervention Strategies to Resolve Exacerbation and Progressive Lung Function Decline in Cystic Fibrosis" Journal of Personalized Medicine 11, no. 2: 96. https://doi.org/10.3390/jpm11020096
APA StyleVij, N. (2021). Prognosis-Based Early Intervention Strategies to Resolve Exacerbation and Progressive Lung Function Decline in Cystic Fibrosis. Journal of Personalized Medicine, 11(2), 96. https://doi.org/10.3390/jpm11020096