
Newly Developed Self-Assembling Antioxidants as Potential Therapeutics for the Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxidative Stress and Cancer
3. Conventional Antioxidants for Potential Cancer Therapy
4. Novel Self-Assembling Antioxidants; Nitroxide Radical-Containing Nanoparticle (RNP)
4.1. Design and Structure of RNPs
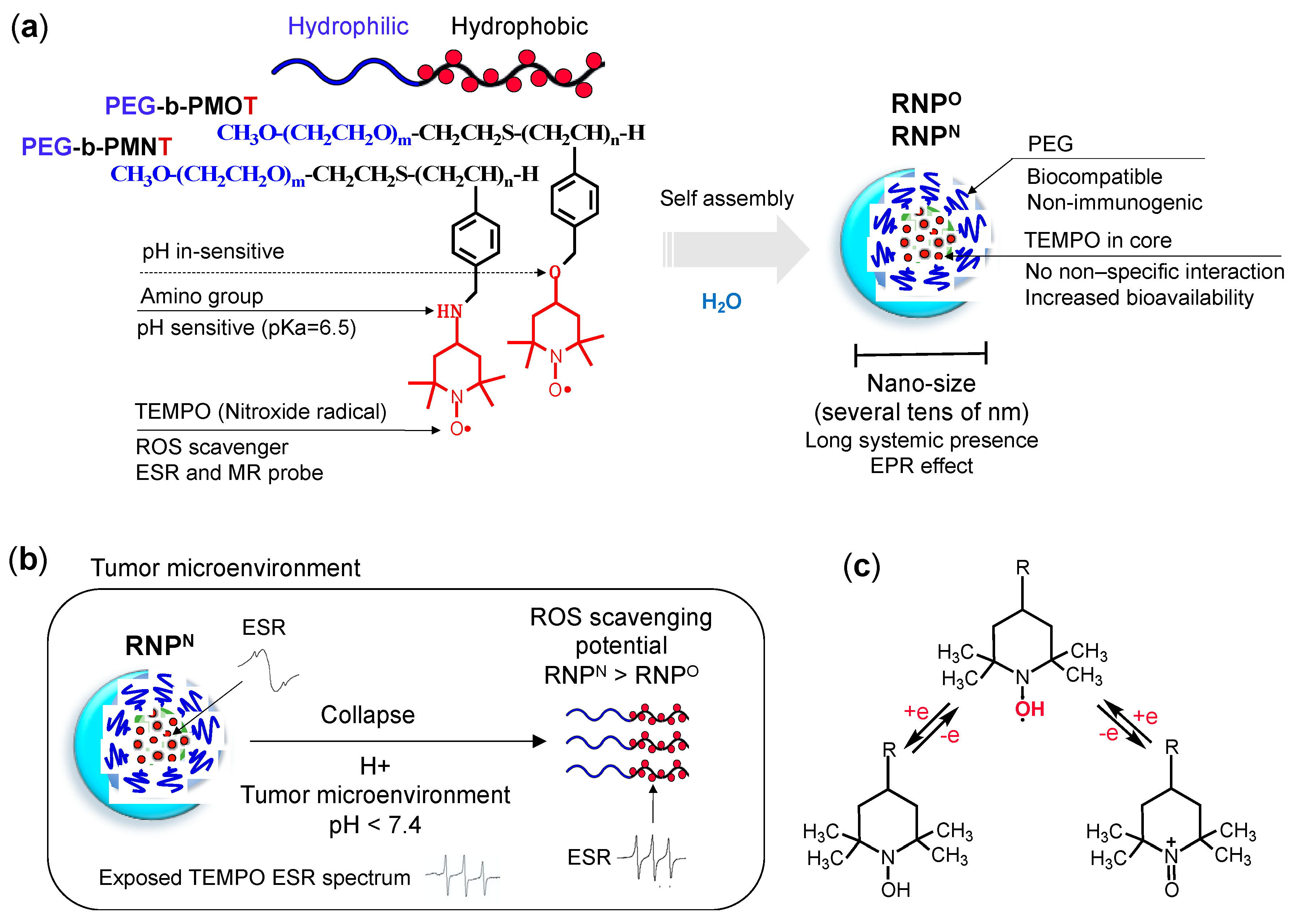
- Structure: RNP, a polymeric micelle made of amphipathic block copolymers, can stably disperse in in vivo harsh conditions due to the entanglement of hydrophobic segments in its core (Figure 2a) [79]. It is reported that PEG imparts biocompatible characteristic to nanoparticle by inhibiting electrostatic and hydrophobic interactions with proteins and cells sterically, thereby increasing the stability of nanoparticle [80]. Unpaired radical in TEMPO is stable by preventing the coupling with each other due to the protection by four methyl groups surrounding it. However, since ROS are small molecular radical species, they rapidly react with TEMPO’s nitroxide radical. Although TEMPO is a highly reactive radical, it is conjugated via covalent linkage in the nanoparticle core; hence, non-specific interaction like LMW TEMPOL can be avoided upon administration. These characteristics potentially improve their accumulation in the target site via the EPR effect, which increases their therapeutic effects and prevents their premature renal excretion.
- Size: Core-shell type polymer micelles with several tens of nanometer in size (20–50 nm) ensures efficient accumulation in target intestinal mucosa (oral administration; colon cancer) or tumor vicinity (intravenous administration: breast cancer), additionally supported by the EPR effect [70,81,82]. It should be noted that the size range of RNP used in various anti-cancer studies, were small enough to prevent activation of the phagocytic system (≤100 nm cutoff size). Conversely, RNPs were large enough to evade rapid renal clearance (≥5.5 nm cutoff size) [83,84].
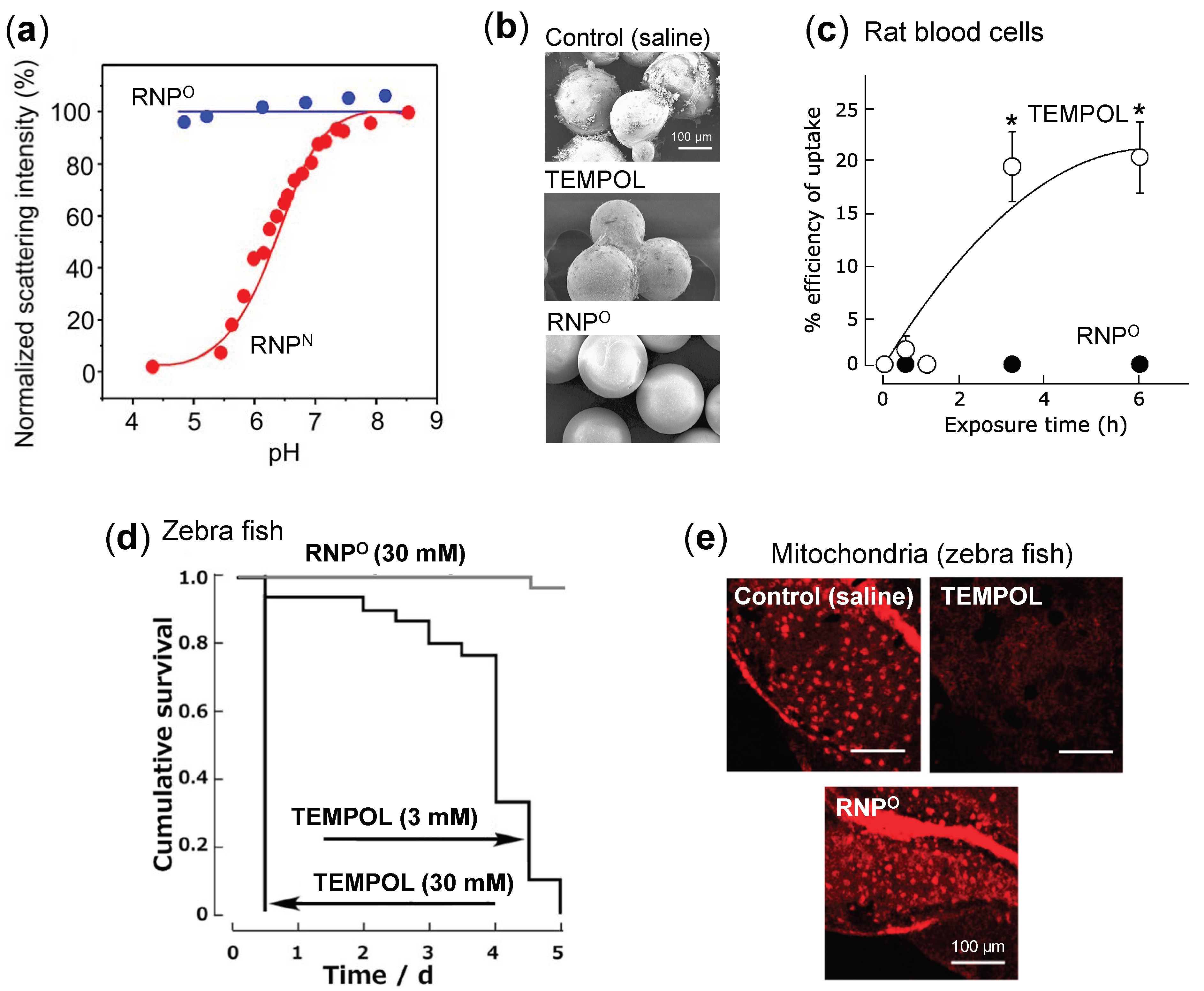
- Stability: Dynamic light scattering studies confirmed that RNPO is stable under various pH 4–8.5, whereas pH-sensitive RNPN was stable at pH 7.4 but decreased with a decrease in pH, confirming its collapse at low pH (diseased condition; tumor) (Figure 3a). Nonetheless, both the micelles maintained structural integrity at physiological pH 7.4, confirming the structural stability in the blood [77]. Furthermore, in ex vivo spiking experiments, we demonstrated that RNP do not internalize in the blood cells and prevents blood cell aggregation on the glass beads, which was in sharp contrast to TEMPOL (Figure 3b,c) [85]. This inert characteristic of RNPs with blood is extremely important for the systemic administration of nanoparticles.
- ESR active properties: ESR measurement shows a characteristics sharp triplet peak of the exposed TEMPO radical (an interaction between 14N nuclei and the unpaired electron), but when confined in the core of RNP, the ESR signal of TEMPO broadens at the same magnetic field, due to restricted mobility of the radicals in RNP’s solid core (Figure 2b) [75]. Due to this characteristic, it is very convenient to confirm the integrity and collapse of RNP and localization of RNPs for the pharmacokinetics studies.
4.2. Safety of RNPs
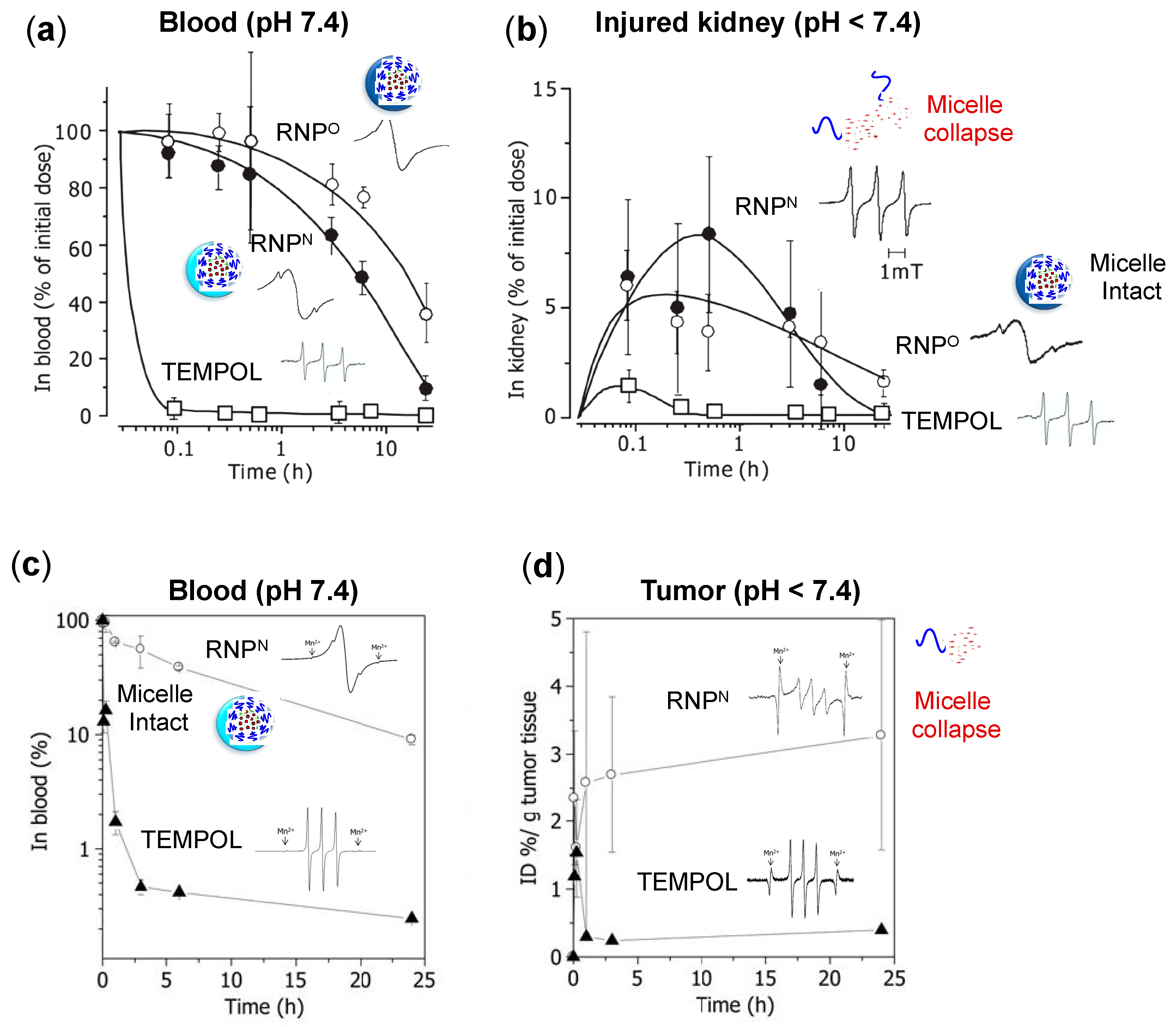
4.3. Pharmacokinetic Properties of RNPs
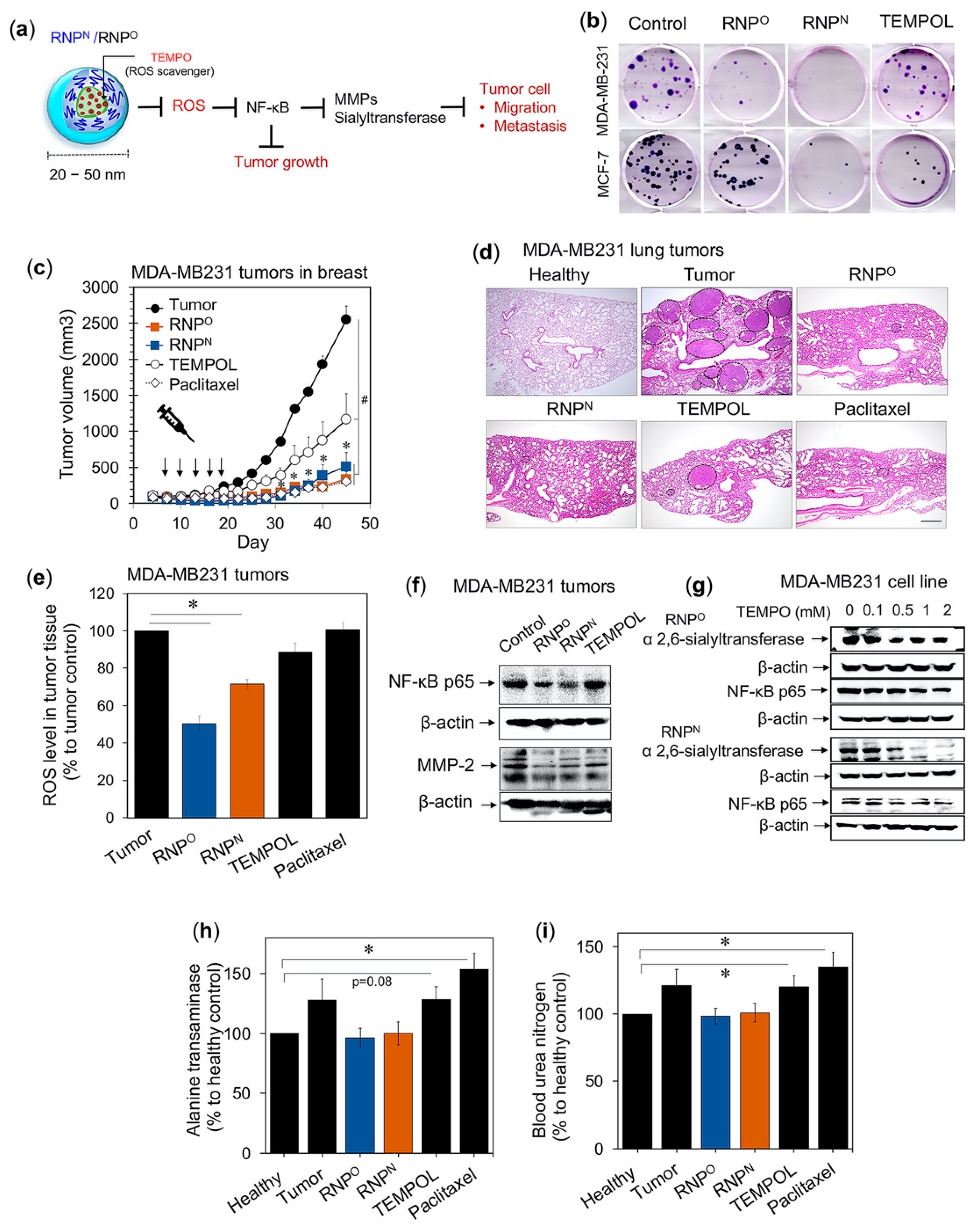
4.4. RNPs for Cancer Therapy
4.4.1. RNPs Inhibit the Tumorigenic Potential of Triple-Negative Breast Cancer
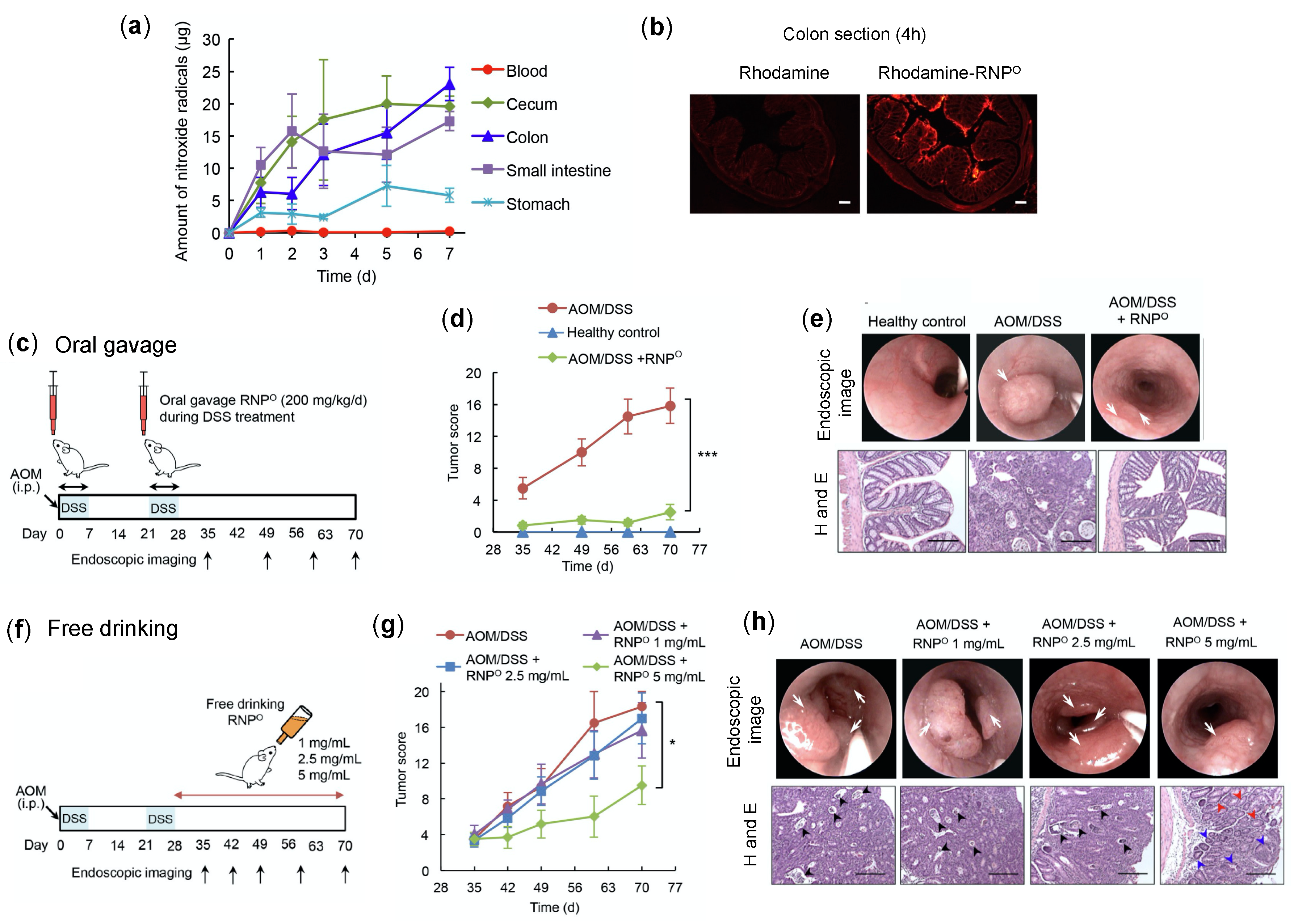
4.4.2. RNPs Inhibit the Tumor Growth and Progression of Colitis-Associated Cancer
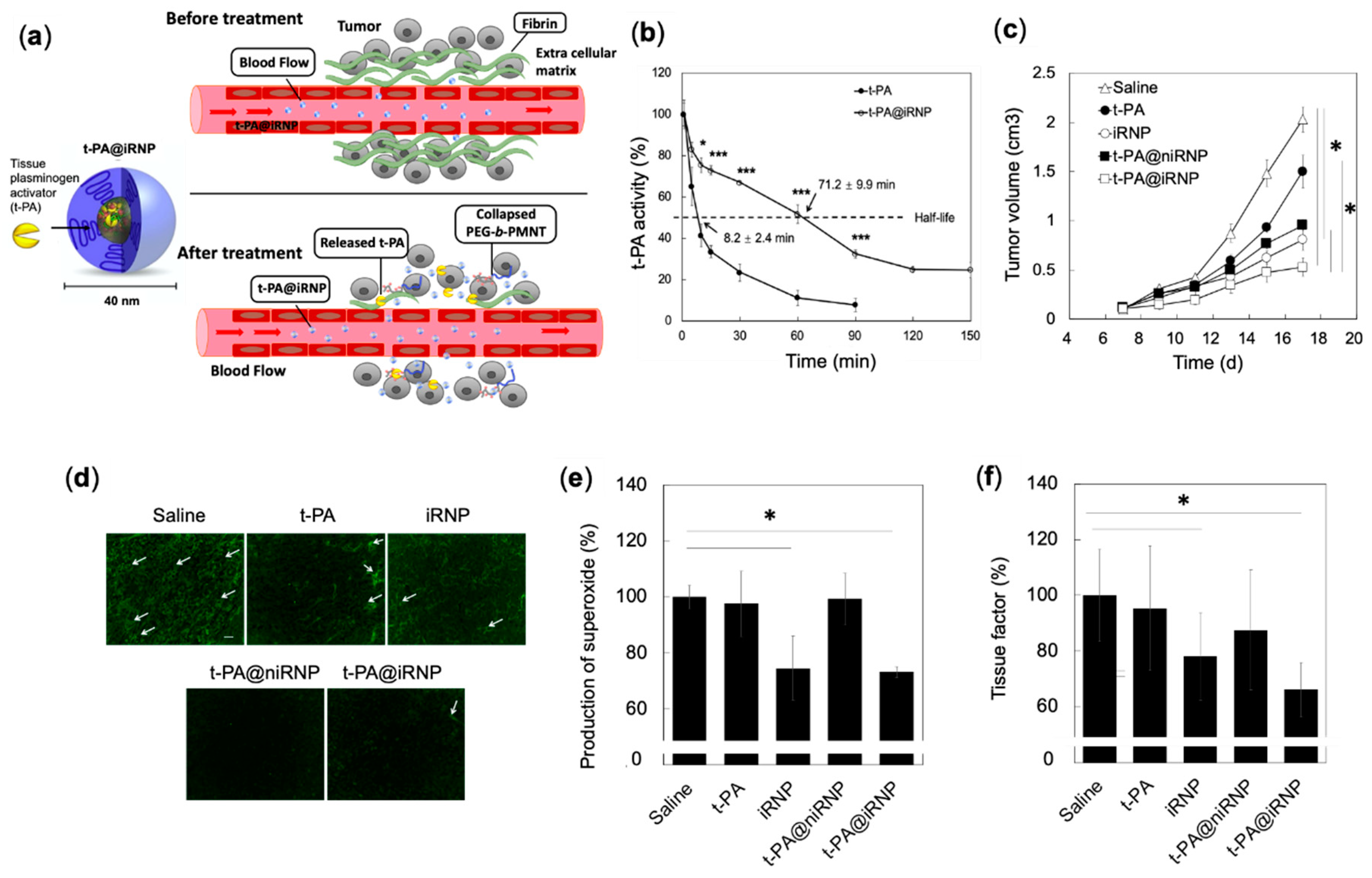
4.4.3. Synergistic Effects of RNP and Fibrinolytic Tissue Plasminogen Activator for Colon Cancer Therapy
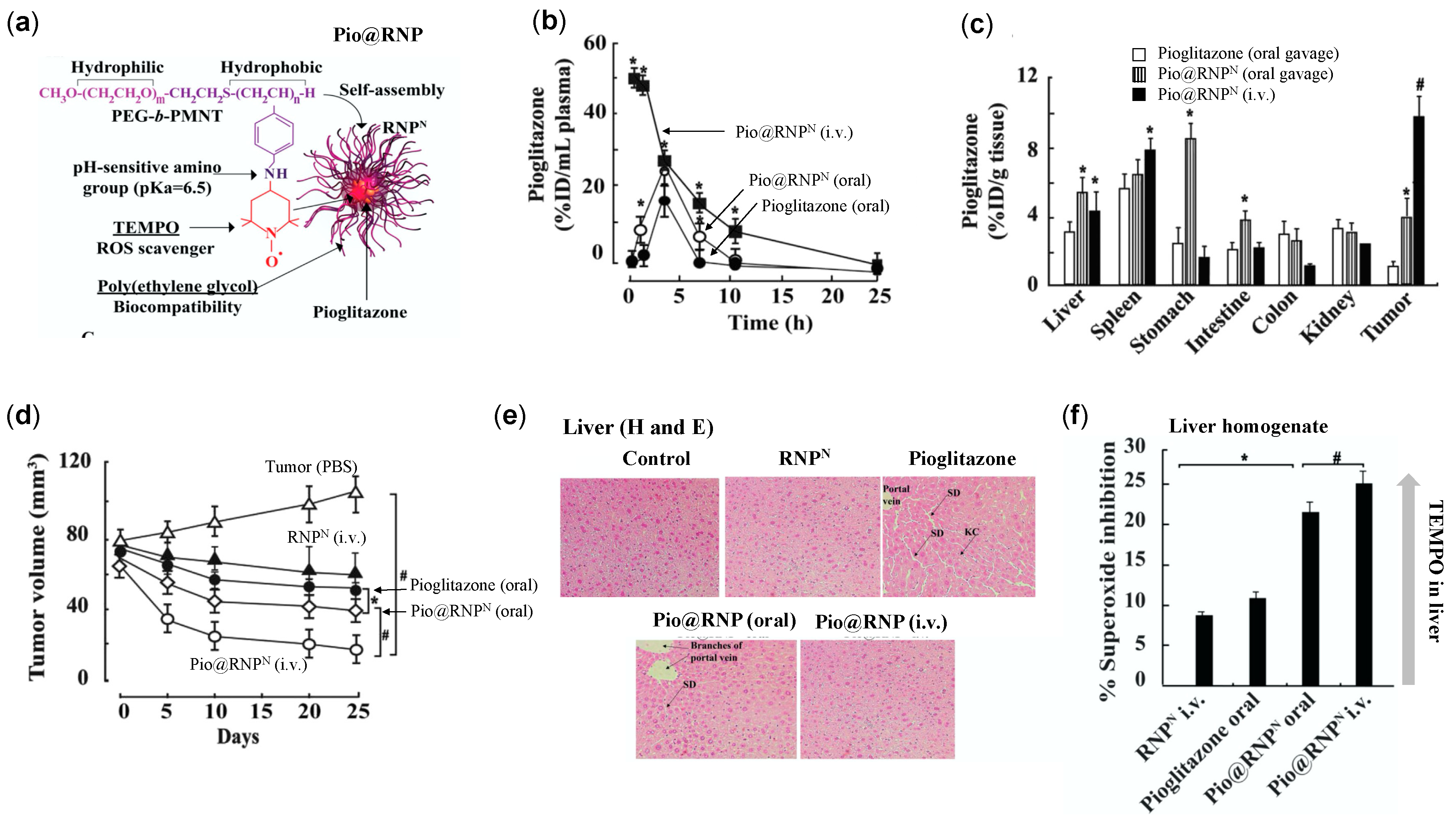
4.4.4. RNPs Enhances the Therapeutic Efficiency of Pioglitazone on Prostate Cancer
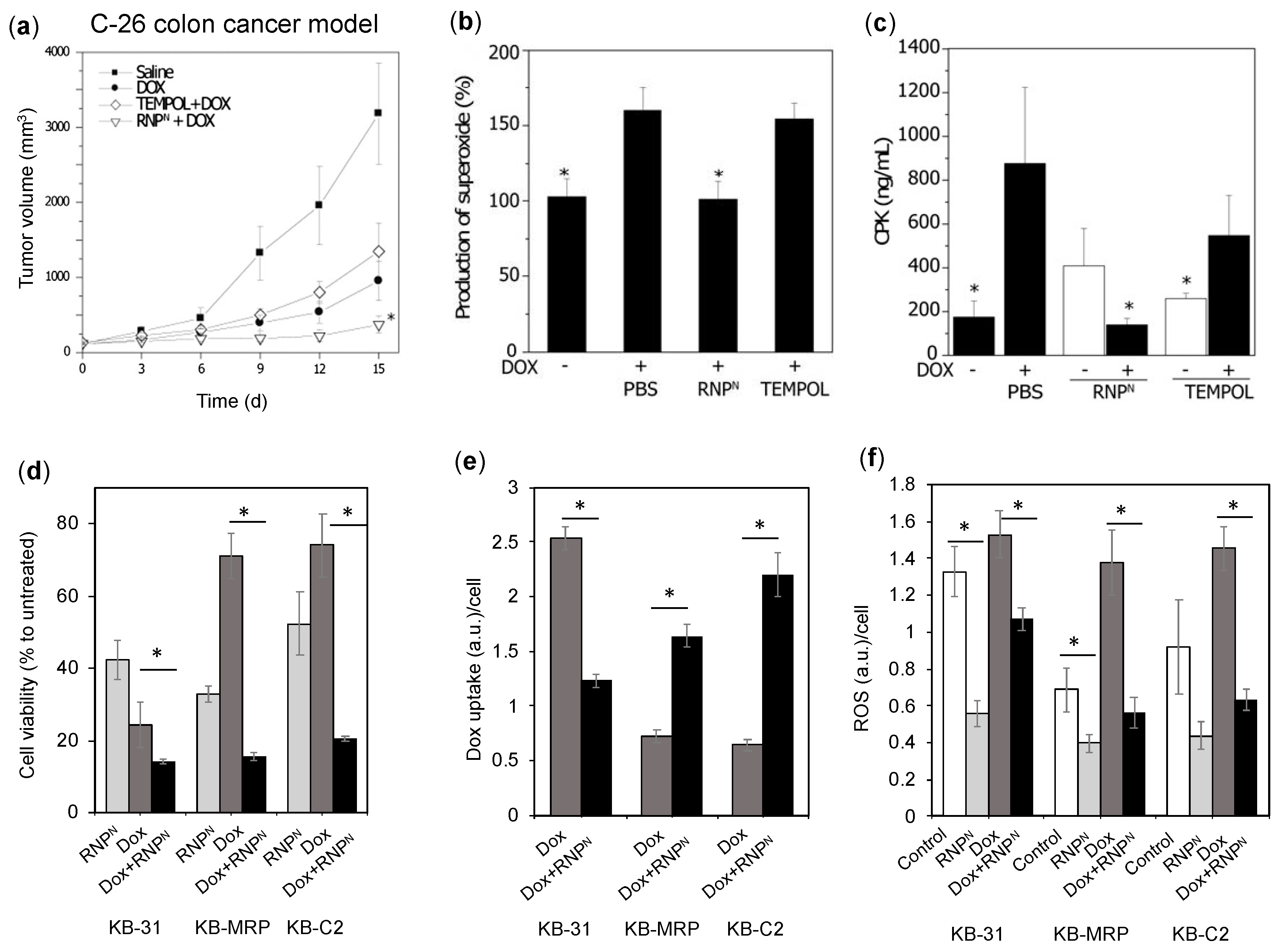
4.4.5. RNPs Enhances the Therapeutic Efficiency of Doxorubicin on Colon Cancer and Epidermoid Cancers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mukherji, S.M.; Singh, S.P. Reaction Mechanism in Organic Chemistry; Macmillan India Press: Chennai, India, 1986. [Google Scholar]
- Gomberg, M. An incidence of trivalent carbon trimethylphenyl. J. Am. Chem. Soc. 1900, 22, 757–771. [Google Scholar] [CrossRef]
- Gerschman, R.; Gilbert, D.L.; Nye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen poisoning and x-irradiation: A mechanism in common. Science 1954, 119, 623–626. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 24, 6049–6055. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and other reactive species in disease. In Nature Encyclopedia of Life Sciences; Nature Publishing Group: London, UK, 2001; pp. 1–7. [Google Scholar]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Dreher, D.; Junod, A.F. Role of oxygen free radicals in cancer development. Eur. J. Cancer 1996, 32, 30–38. [Google Scholar] [CrossRef]
- Fuchs-Tarlovsky, V. Role of antioxidants in cancer therapy. Nutrition 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox. Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Zazzo, E.D.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 202052, 192–203. [Google Scholar] [CrossRef]
- Hsie, A.W.; Recio, L.; Katz, D.S.; Lee, C.Q.; Wagner, M.; Schenley, R.L. Evidence for reactive oxygen species inducing mutations in mammalian cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9616–9620. [Google Scholar] [CrossRef]
- Weiss, S.J. Tissue destruction by neutrophils. N. Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [PubMed]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Gimeno, C.J.; Ames, B.N. Urinary 8-hydroxy- 2-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc. Natl. Acad. Sci. USA 1989, 86, 9697–9701. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Ames, B.N. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science 1983, 221, 1256–1264. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Xu, W.; Trepel, J.; Neckers, L. Ras, ROS and proteotoxic stress: A delicate balance. Cancer Cell 2011, 20, 281–282. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Pervaiz, S. Redox regulation of p53, redox effectors regulated by p53: A subtle balance. Antioxid. Redox Signal. 2012, 16, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Simon, H.U. A novel link between p53 and ROS. Cell Cycle 2013, 12, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Pisconti, S.; Della Vittoria Scarpati, G. P53 mutations and cancer: A tight linkage. Ann. Transl. Med. 2016, 4, 522. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. p53 function and dysfunction. Cell 1992, 70, 523–526. [Google Scholar] [CrossRef]
- Amiri, K.I.; Richmond, A. Role of nuclear factor-kappa B in melanoma. Cancer Metast. Rev. 2005, 24, 301–313. [Google Scholar] [CrossRef]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef]
- Knight, J.A. Free radicals, antioxidants, and the immune system. Ann. Clin. Lab. Sci. 2000, 30, 145–158. [Google Scholar]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. OncoTargets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Larsson, R.; Cerutti, P. Translocation and enhancement of phosphotransferase activity of protein kinase C following exposure in mouse epidermal cells to oxidants. Cancer Res. 1989, 49, 5627–5632. [Google Scholar] [PubMed]
- Gopalakrishna, R.; Anderson, W. Ca2+-and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc. Natl. Acad. Sci. USA 1989, 86, 6758–6762. [Google Scholar] [CrossRef]
- Lopez-Ilasaca, M.; Crespo, P.; Pellici, P.G.; Gutkind, J.S.; Wetzker, R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science 1997, 275, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Jiang, B.H.; Leonard, S.S.; Corum, L.; Zhang, Z.; Roberts, J.R.; Antonini, J.; Zheng, J.Z.; Flynn, D.C.; Castranova, V.; et al. p38 Signaling-mediated hypoxia-inducible factor 1 alpha and vascular endothelial growth factor induction by Cr (VI) in DU145 human prostate carcinoma cells. J. Biol. Chem. 2002, 277, 45041–45048. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.C.; Young, M.R.; Cmarik, J.; Colburn, N.H. Activator protein 1 (AP-1)- and nuclear factor kappa B (NF-kappa B)- dependent transcriptional events in carcinogenesis. Free Rad. Biol. Med. 2000, 28, 1338–1348. [Google Scholar] [CrossRef]
- Huang, C.; Li, J.; Costa, M.; Zhang, Z.; Leonard, S.S.; Castranova, V.; Vallyathan, V.; Ju, G.; Shi, X. Hydrogen peroxide mediates activation of nuclear factor of activated T cells (NFAT) by nickel sulfide. Cancer Res. 2001, 61, 8051–8057. [Google Scholar] [PubMed]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer. 2009, 9, 810–820. [Google Scholar] [CrossRef]
- Um, H.D. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: A review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 2016, 7, 5193–5203. [Google Scholar] [CrossRef]
- Sharma, M.; Rajappa, M.; Kumar, G.; Sharma, A. Oxidant-antioxidant status in Indian patients with carcinoma of posterior one-third of tongue. Cancer Biomark. 2009, 5, 253–260. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, D.M.; Lee, C.H.; Heo, S.H.; Won, S.Y.; Im, J.H.; Cho, M.K.; Nam, H.S.; Lee, S.H. Suppression of human prostate cancer PC-3 cell growth by N-acetylcysteine involves over-expression of Cyr61. Toxicol. In Vitro 2011, 25, 199–205. [Google Scholar] [CrossRef]
- Lee, M.F.; Chan, C.Y.; Hung, H.C.; Chou, I.T.; Yee, A.S.; Huang, C.Y. N-acetylcysteine (NAC) inhibits cell growth by mediating the EGFR/Akt/HMG box-containing protein 1 (HBP1) signaling pathway in invasive oral cancer. Oral Oncol. 2013, 49, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, C.; Fang, T.; Li, T.; Lv, G.; Han, Q.; Yang, W.; Wang, H. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol. 2018, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Wang, Q.M.; Feng, L.Y.; Ke, Y.D.; Xu, Q.Z.; Wei, A.Y.; Zhang, C.; Ying, R.B. High-dose vitamin C suppresses the invasion and metastasis of breast cancer cells via inhibiting epithelial-mesenchymal transition. OncoTargets Ther. 2019, 12, 7405–7413. [Google Scholar] [CrossRef] [PubMed]
- Turley, J.M.; Fu, T.; Ruscetti, F.W.; Mikovits, J.A.; Bertolette, D.C., 3rd; Birchenall-Roberts, M.C. Vitamin E succinate induces Fas-mediated apoptosis in estrogen receptor-negative human breast cancer cells. Cancer Res. 1997, 57, 881–890. [Google Scholar] [PubMed]
- Weiping, Y.; Qiao, Y.L.; Feras, M.; Hantash, B.G. Sanders and Kimberly Kline. Activation of extracellular signal-regulated kinase and c-Jun-NH2-terminal kinase but not p38 mitogen-activated protein kinases is required for RRR-α-Tocopheryl succinate-induced apoptosis of human breast cancer cells. Cancer Res. 2001, 61, 6569–6576. [Google Scholar]
- Choi, J.A.; Kim, J.Y.; Lee, J.Y.; Kang, C.M.; Kwon, H.J.; Yoo, Y.D.; Kim, T.W.; Lee, Y.S.; Lee, S.J. Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin. Int. J. Oncol. 2001, 19, 837–844. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Budhraja, A.; Son, Y.O.; Wang, X.; Zhang, Z.; Ding, S.; Wang, L.; Hitron, A.; Lee, J.C.; Xu, M.; et al. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR- 2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS ONE 2012, 7, e47516. [Google Scholar] [CrossRef]
- Richter, M.; Ebermann, R.; Marian, B. Quercetin-induced apoptosis in colorectal tumor cells: Possible role of EGF receptor signaling. Nutr. Cancer 1999, 34, 88–99. [Google Scholar] [CrossRef]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef]
- Münzel, T.; Afanas’ev, I.B.; Kleschyov, A.L.; Harrison, D.G. Detection of superoxide in vascular tissue. Arter. Thromb. Vasc. Biol. 2002, 22, 1761–1768. [Google Scholar] [CrossRef]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007, 42, 1632–1650. [Google Scholar] [CrossRef] [PubMed]
- Suy, S.; Mitchell, J.B.; Ehleiter, D.; Haimovitz-Friedman, A.; Kasid, U. Nitroxides tempol and tempo induce divergent signal transduction pathways in MDA-MB 231 breast cancer cells. J. Biol. Chem. 1998, 273, 17871–17878. [Google Scholar] [CrossRef] [PubMed]
- Gariboldi, M.B.; Lucchi, S.; Caserini, C.; Supino, R.; Oliva, C.; Monti, E. Antiproliferative effect of the piperidine nitroxide TEMPOL on neoplastic and nonneoplastic mammalian cell lines. Free Radic. Biol. Med. 1998, 24, 913–923. [Google Scholar] [CrossRef]
- Gariboldi, M.B.; Rimoldi, V.; Supino, R.; Favini, E.; Monti, E. The nitroxide tempol induces oxidative stress, p21(WAF1/CIP1), and cell death in HL60 cells. Free Radic. Biol. Med. 2000, 29, 633–641. [Google Scholar] [CrossRef]
- Gariboldi, M.B.; Ravizza, R.; Petterino, C.; Castagnaro, M.; Finocchiaro, G.; Monti, E. Study of in vitro and in vivo effects of the piperidine nitroxide Tempol-a potential new therapeutic agent for gliomas. Eur. J. Cancer 2003, 39, 829–837. [Google Scholar] [CrossRef]
- Schubert, R.; Erker, L.; Barlow, C.; Yakushiji, H.; Larson, D.; Russo, A.; Mitchell, J.B.; Wynshaw-Boris, A. Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Hum. Mol. Genetics 2004, 13, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Xavier, S.; DeLuca, A.M.; Sowers, A.L.; Cook, J.A.; Krishna, M.C.; Hahn, S.M.; Russo, A.A. A low molecular weight antioxidant decreases weight and lowers tumor incidence. Free Rad. Biol. Med. 2003, 34, 93–102. [Google Scholar] [CrossRef]
- Suy, S.; Mitchell, J.B.; Samuni, A.; Mueller, S.; Kasid, U. Nitroxide tempo, a small molecule, induces apoptosis in prostate carcinoma cells and suppresses tumor growth in athymic mice. Cancer 2005, 103, 1302–1313. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M., Jr.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The selenium and vitamin E cancer prevention trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Barnett, M.J.; Kristal, A.R.; Ambrosone, C.B.; King, I.B.; Thornquist, M.; Goodman, G.G. Dietary supplement use and prostate cancer risk in the carotene and retinol efficacy trial. Cancer Epidemiol. Biomark. 2009, 18, 2202–2206. [Google Scholar] [CrossRef] [PubMed]
- Blot, W.J.; Li, J.Y.; Taylor, P.R.; Guo, W.; Dawsey, S.; Wang, G.Q.; Yang, C.S.; Zheng, S.F.; Gail, M.; Li, G.Y.; et al. Nutrition intervention trials in Linxian, China: Supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J. Natl. Cancer Inst. 1993, 85, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Meyskens, F.L., Jr.; Omenn, G.S.; Valanis, B.; Williams, J.H., Jr. The beta-carotene and retinol efficacy rrial: Incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J. Natl. Cancer Inst. 2004, 96, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen consumption and usage during physical exercise: The balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, S. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Gal, L.K.; Ibrahim, M.; Wiel, C.; Sayin, V.; Akula, M.; Karlsson, C. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; Saedeleer, C.J.; Danhier, P.A.; Copetti, T.; Suveera, D.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. Mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Rezaei-Sadabady, R.; Eidi, A.; Zarghami, N.; Barzegar, A. Intracellular ROS protection efficiency and free radical-scavenging activity of quercetin and quercetin-encapsulated liposomes. Artif. Cells Nanomed. Biotechnol. 2016, 44, 128–134. [Google Scholar] [CrossRef]
- Hijaz, M.; Das, S.; Mert, I.; Gupta, A.; Al-Wahab, Z.; Tebbe, C.; Sajad, D.; Chinna, J.; Giri, S.; Munkarah, A.; et al. Folic acid tagged nanoceria as a novel therapeutic agent in ovarian cancer. BMC Cancer 2016, 16, 220. [Google Scholar] [CrossRef]
- Morry, J.; Ngamcherdtrakul, W.; Gu, S.; Reda, M.; Castro, D.J.; Sangvanich, T.; Gray, J.W.; Yantasee, W. Targeted treatment of metastatic breast cancer by PLK1 siRNA delivered by an antioxidant nanoparticle platform. Mol. Cancer Ther. 2017, 16, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, T.; Fu, C.; Tan, L.; Meng, X.; Liu, H. Biodistribution, excretion, and toxicity of mesoporous silica nanoparticles after oral administration depend on their shape. Nanomedicine 2015, 11, 1915–1924. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, T.; Nagasaki, Y. Reactive oxygen species-scavenging nanomedicines for the treatment of oxidative stress injuries. Adv. Healthc. Mater. 2014, 3, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, T.; Nagasaki, Y. Nitroxyl radical-containing nanoparticles for novel nanomedicine against oxidative stress injury. Nanomedicine 2011, 6, 509–518. [Google Scholar] [CrossRef]
- Yoshitomi, T.; Hirayama, A.; Nagasaki, Y. The ROS scavenging and renal protective effects of pH-responsive nitroxide radical-containing nanoparticles. Biomaterials 2011, 32, 8021–8028. [Google Scholar] [CrossRef]
- Yoshitomi, T.; Suzuki, R.; Mamiya, T.; Matsui, H.; Hirayama, A.; Nagasaki, Y. pH-Sensitive radical-containing-nanoparticle (RNP) for the L-Band-EPR imaging of low pH circumstances. Bioconjugate Chem. 2009, 20, 1792–1798. [Google Scholar] [CrossRef]
- Francis, M.; Cristea, M.; Winnik, F. Polymeric micelles for oral drug delivery: Why and how. Pure Appl. Chem. 2004, 76, 1321–1335. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Sha, S.; Vong, L.B.; Chonpathompikunlert, P.; Yoshitomi, T.; Matsui, H.; Nagasaki, Y. Suppression of NSAID-induced small intestinal inflammation by orally administered redox nanoparticles. Biomaterials 2013, 34, 8393–8400. [Google Scholar] [CrossRef]
- Yoshitomi, T.; Ozaki, Y.; Thangavel, S.; Nagasaki, Y. Redox nanoparticle therapeutics to cancer—Increase in therapeutic effect of doxorubicin, suppressing its adverse effect. J. Control. Release 2013, 172, 137–143. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Ipe, B.I.; Bawendi, M.G.; Frangioni, J. Renal clearance of nanoparticles. Nat. Biotechnol. 2017, 25, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Huang, L. Pharmacokinetics and biodistribution of nanoparticles. Mol. Pharm. 2008, 5, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Yoshitomi, T.; Nagasaki, Y. The behavior of ROS scavenging nanoparticles in blood. J. Clin. Biochem. 2014, 54, 166–173. [Google Scholar] [CrossRef][Green Version]
- Nagasaki, Y. Design and application of redox polymers for nanomedicine. Polym. J. 2018, 50, 821–836. [Google Scholar] [CrossRef]
- Monti, E.; Supino, R.; Colleoni, M.; Costa, B.; Ravizza, R.; Gariboldi, M.B. Nitroxide TEMPOL impairs mitochondrial function and induces apoptosis in HL60 cells. J. Cell. Biochem. 2001, 82, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.B.; Kobayashi, M.; Nagasaki, Y. Evaluation of the toxicity and antioxidant activity of redox nanoparticles in Zebrafish (Danio rerio) embryos. Mol. Pharm. 2016, 13, 3091–3097. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.M.; Tochner, Z.; Krishna, C.M.; Glass, J.; Wilson, L.; Samuni, A.; Sprague, M.; Venzon, D.; Glatstein, E.; Mitchell, J.B.; et al. Tempol, a stable free radical, is a novel murine radiation protector. Cancer Res. 1992, 52, 1750–1753. [Google Scholar]
- Magalhães, P.A.; de Brito, T.S.; Freire, R.S.; da Silva, M.T.; dos Santos, A.A.; Vale, M.L.; de Menezes, D.B.; Martins, A.M.; Libório, A.B. Metabolic acidosis aggravates experimental acute kidney injury. Life Sci. 2016, 146, 58–65. [Google Scholar] [CrossRef]
- Azamjah, N.; Soltan-Zadeh, Y.; Zayeri, F. Global trend of breast cancer mortality rate: A 25-Year Study. Asian Pac. J. Cancer Prev. 2019, 20, 2015–2020. [Google Scholar] [CrossRef]
- Yeh, C.C.; Hou, M.F.; Tsai, S.M.; Lin, S.K.; Hsiao, J.K.; Huang, J.C.; Wang, L.H.; Wu, S.H.; Hou, L.A.; Ma, H.; et al. Superoxide anion radical, lipid peroxides and antioxidant status in the blood of patients with breast cancer. Clin. Chim. Acta 2005, 361, 101–111. [Google Scholar] [CrossRef]
- Renschler, F. The emerging role of reactive oxygen species in cancer therapy. Eur. J. Cancer 2004, 40, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Kumaraguruparan, R.; Subapriya, R.; Kablimoorthy, J.; Nagini, S. Antioxidant profile in the circulation of patients with fibroadenoma and adenocarcinoma of the breast. Clin. Biochem. 2002, 35, 275–279. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; De Marco, P.; Cirillo, F.; Cappello, A.R.; Dolce, V.; Belfiore, A.; et al. Copper activates HIF-1alpha/GPER/VEGF signalling in cancer cells. Oncotarget 2015, 6, 34158–34177. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Coleman, M.C.; Walsh, S.A.; Spitz, D.R.; Goswami, P.C. Differential susceptibility of nonmalignant human breast epithelial cells and breast cancer cells to thiol antioxidant-induced G(1)-delay. Antioxid. Redox Signal. 2005, 7, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Zhang, S.; Fulton, A.M. Sublethal oxidative stress inhibits tumor cell adhesion and enhances experimental metastasis of murine mammary carcinoma. Clin. Exp. Metastasis 1995, 13, 16–22. [Google Scholar] [CrossRef]
- Shashni, B.; Nagasaki, Y. Nitroxide radical-containing nanoparticles attenuate tumorigenic potential of triple negative breast cancer. Biomaterials 2018, 178, 48–62. [Google Scholar] [CrossRef]
- Shashni, B.; Horiguchi, Y.; Kurosu, K.; Furusho, H.; Nagasaki, Y. Application of surface enhanced Raman spectroscopy as a diagnostic system for hypersialylated metastatic cancers. Biomaterials 2017, 134, 143–153. [Google Scholar] [CrossRef]
- Duffy, M.J.; Maguire, T.M.; Hill, A.; McDermott, E.; Higgins, N.O. Metal-loproteinases: Role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000, 2, 252–257. [Google Scholar] [CrossRef]
- Bull, C.; Stoel, M.A.; Brok, M.H.D.; Adema, G.J. Sialic acids sweeten a tumor’s life. Cancer Res. 2014, 74, 3199–3204. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Gommeaux, J.; Cano, C.; Garcia, S.; Gironella, M.; Pietri, S.; Culcasi, M.; Gommeaux, J.; Pébusque, M.J.; Malissen, B.; Dusetti, N.; et al. Colitis and colitis-associated cancer are exacerbated in mice deficient for tumor protein 53-induced nuclear protein 1. Mol. Cell. Biol. 2007, 27, 2215–2228. [Google Scholar] [CrossRef] [PubMed]
- Tenesa, A.; Dunlop, M.G. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat. Rev. Genet. 2009, 10, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.B.; Yoshitomi, T.; Matsui, H.; Nagasaki, Y. Development of an oral nanotherapeutics using redox nanoparticles for treatment of colitis-associated colon cancer. Biomaterials 2015, 55, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.B.; Tomita, T.; Yoshitomi, T.; Matsui, H.; Nagasaki, Y. An orally administered redox nanoparticle that accumulates in the colonic mucosa and reduces colitis in mice. Gastroenterology 2012, 143, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar]
- Mars, W.M.; Zarnegar, R.; Michalopoulos, G.K. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am. J. Pathol. 1993, 143, 949–958. [Google Scholar]
- Zhang, B.; Jiang, T.; She, X.; Shen, S.; Wang, S.; Deng, J.; Shi, W.; Mei, H.; Hu, Y.; Pang, Z.; et al. Fibrin degradation by rtPA enhances the delivery of nanotherapeutics to A549 tumors in nude mice. Biomaterials 2016, 96, 63–71. [Google Scholar] [CrossRef]
- Nilsson, T.; Wallen, P.; Mellbring, G. In vivo metabolism of human tissue-type plasminogen activator. Scand. J. Haematol. 1984, 33, 49–53. [Google Scholar] [CrossRef]
- Mei, T.; Shashni, B.; Maeda, H.; Nagasaki, Y. Fibrinolytic tissue plasminogen activator installed redox-active nanoparticles (t-PA@iRNP) for cancer therapy. Biomaterials 2020, 259, 120290. [Google Scholar] [CrossRef]
- Mei, T.; Kim, A.; Vong, L.B.; Marushima, A.; Puentes, S.; Matsumaru, Y.; Matsumura, A.; Nagasaki, Y. Encapsulation of tissue plasminogen activator in pH-sensitive self-assembled antioxidant nanoparticles for ischemic stroke treatment-Synergistic effect of thrombolysis and antioxidant. Biomaterials 2019, 215, 119209. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.M.; Patierno, S.R.; Rickles, F.R. Tissue factor and fibrin in tumor angiogenesis. Semin. Thromb. Hemost. 2004, 30, 31–44. [Google Scholar] [PubMed]
- Cadroy, Y.; Dupouy, D.; Boneu, B.; Plaisancié, H. Polymorphonuclear leukocytes modulate tissue factor production by mononuclear cells: Role of reactive oxygen species. J. Immunol. 2000, 164, 3822–3828. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Yoshitomi, T.; Sakharkar, M.K.; Nagasaki, Y. Redox nanoparticle increases the chemotherapeutic efficiency of pioglitazone and suppresses its toxic side effects. Biomaterials 2016, 99, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Floyd, J.S.; Barbehenn, E.; Lurie, P.; Wolfe, S.M. Case series of liver failure associated with rosiglitazone and pioglitazone. Pharmaco epidemiol. Drug Saf. 2009, 18, 1238–1243. [Google Scholar]
- Rabbani, S.I.; Devi, K.; Khanam, S. Role of pioglitazone with metformin or glimepiride on oxidative stress-induced nuclear damage and reproductive toxicity in diabetic rats. Malays. J. Med. Sci. 2010, 17, 3–11. [Google Scholar]
- Fojo, T.; Menefee, M. Mechanisms of multidrug resistance: The potential role of microtubule-stabilizing agents. Ann. Oncol. 2007, 18, v3–v8. [Google Scholar] [CrossRef]
- Holohan, C.; Schaeybroeck, S.V.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Saeki, T.; Nomizu, T.; Toi, M.; Ito, Y.; Noguchi, S.; Kobayashi, T.; Asaga, T.; Minami, H.; Yamamoto, N.; Aogi, K.; et al. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J. Clin. Oncol. 2007, 25, 411–417. [Google Scholar] [CrossRef]
- Friedenberg, W.R.; Rue, M.; Blood, E.A.; Dalton, W.S.; Shustik, C.; Larson, R.A.; Sonneveld, P.; Greipp, P.R. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): A trial of the Eastern Cooperative Oncology Group. Cancer 2006, 106, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef] [PubMed]
- Bentires-Alj, M.; Barbu, V.; Fillet, M.; Chariot, A.; Relic, B.; Jacobs, N.; Gielen, J.; Merville, M.P.; Bours, B. NF-κB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 2003, 22, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E. Implications of anthracycline-resistant and taxane-resistant meta- static breast cancer and new therapeutic options. Breast J. 2010, 16, 252–263. [Google Scholar] [CrossRef]
- Kokura, S.; Yoshida, N.; Sakamoto, N.; Ishikawa, T.; Takagi, T.; Higashihara, H.; Nakabe, N.; Handa, O.; Naito, Y.; Yoshikawa, T. The radical scavenger edaravone enhances the anti-tumor effects of CPT-11 in murine colon cancer by increasing apoptosis viainhibition of NF-kappaB. Cancer Lett. 2005, 229, 223–233. [Google Scholar] [CrossRef] [PubMed]
- DeAtley, S.M.; Aksenov, M.Y.; Aksenova, M.V.; Jordan, B.; Carney, J.M.; Butterfield, D.A. Adriamycin-induced changes of creatine kinase activity in vivo and in cardiomyocyte culture. Toxicology 1999, 134, 51–62. [Google Scholar] [CrossRef]
- Shashni, B.; Alshwimi, A.; Minami, M.; Furukawa, T.; Nagasaki, T. Nitroxide radical-containing nanoparticles as potential candidates for overcoming drug resistance in epidermoid cancers. Polymer 2017, 116, 429–438. [Google Scholar] [CrossRef]
- Moriyama, M.; Metzger, S.; van der Vlies, A.J.; Uyama, H.; Ehrbar, M.; Hasegawa, U. Inhibition of angiogenesis by antioxidant micelles. Adv. Healthc. Mater. 2015, 4, 569–575. [Google Scholar] [CrossRef]
- Rocha, S.; Generalov, R.; Pereira Mdo, C.; Peres, I.; Juzenas, P.; Coelho, M.A. Epigallocatechin gallate-loaded polysaccharide nanoparticles for prostate cancer chemoprevention. Nanomedicine 2011, 6, 79–87. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shashni, B.; Nagasaki, Y. Newly Developed Self-Assembling Antioxidants as Potential Therapeutics for the Cancers. J. Pers. Med. 2021, 11, 92. https://doi.org/10.3390/jpm11020092
Shashni B, Nagasaki Y. Newly Developed Self-Assembling Antioxidants as Potential Therapeutics for the Cancers. Journal of Personalized Medicine. 2021; 11(2):92. https://doi.org/10.3390/jpm11020092
Chicago/Turabian StyleShashni, Babita, and Yukio Nagasaki. 2021. "Newly Developed Self-Assembling Antioxidants as Potential Therapeutics for the Cancers" Journal of Personalized Medicine 11, no. 2: 92. https://doi.org/10.3390/jpm11020092
APA StyleShashni, B., & Nagasaki, Y. (2021). Newly Developed Self-Assembling Antioxidants as Potential Therapeutics for the Cancers. Journal of Personalized Medicine, 11(2), 92. https://doi.org/10.3390/jpm11020092