
MET Gene Dysregulation as a Promising Therapeutic Target in Lung Cancer—A Review


Abstract
1. Introduction
2. Testing of MET Gene Abnormalities
3. MET Gene Abnormalities in NSCLC Patients
4. HGFR Inhibitors in Treatment of Patients with EGFR TKI Resistance and MET Gene Amplification
5. Treatment of Patients with METex14 Mutations
6. Other MET Inhibitors in the Treatment of NSCLC Patients with METex14 Mutations and other MET Gene Abnormalities
6.1. Capmatinib
6.2. Tepotinib
6.3. Cabozantinib
6.4. Glesatinib
6.5. Bozitinib
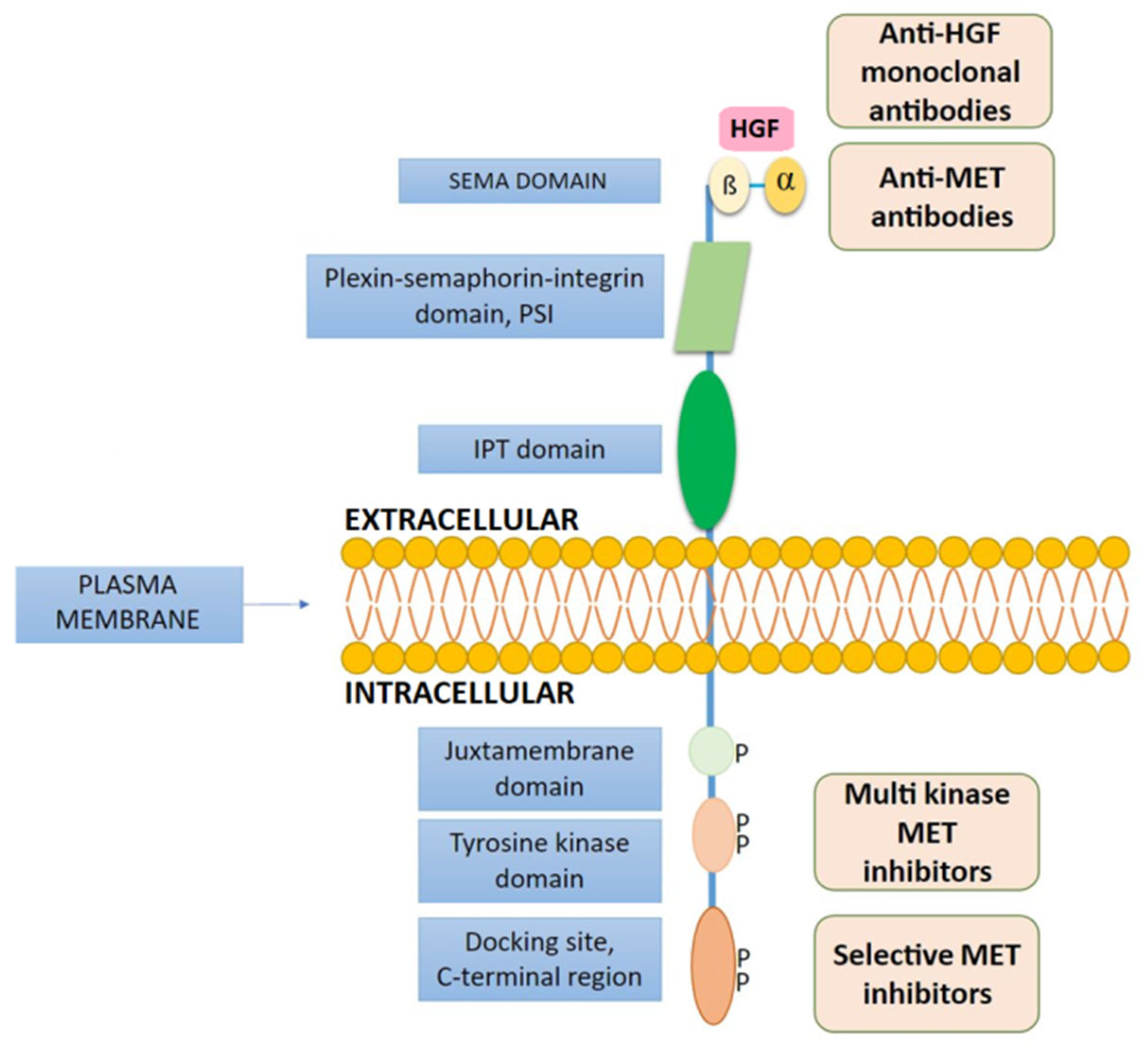
6.6. Anti-MET Monoclonal Antibodies and Immunotherapy in NSCLC Patients with MET Abnormalities
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Drilon, A.; Clark, J.; Weiss, J.; Ou, S.; Camidge, D.R.; Solomon, B.; Otterson, G.; Villaruz, L.; Riely, G.; Heist, R.; et al. Updated antitumor activity of crizotinib in patients with MET exon 14-altered advanced non-small cell lung cancer. J. Thorac. Oncol. 2018, 13, 348. [Google Scholar] [CrossRef]
- O’Brien, O.; Wright, M.C.; O’Brien, C.; Geoghegan, O.; Leonard, N.; Nicholson, S.; Cuffe, S.; Fabre, A.; Jochum, W.; Joerger, M.; et al. Cost-efficient and easy to perform PCR-based assay to identify Met exon 14 skipping in formalin-fixed paraffin-embedded (FFPE) non-small cell lung cancer (NSCLC) samples. Diagnostics 2019, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Berry, L.D.; Aisner, D.L.; Sheren, J.; Boyle, T.; Bunn, P.A., Jr.; Johnson, B.E.; Kwiatkowski, D.J.; Drilon, A.; Sholl, L.M.; et al. MET IHC is a poor screen for MET amplification or MET exon 14 mutations in lung adenocarcinomas: Data from a tri-institutional cohort of the lung cancer mutation consortium. J. Thorac. Oncol. 2019, 14, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: Guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Crepaldi, T.; Pollack, A.L.; Prat, M.; Zborek, A.; Mostov, K.; Comoglio, P.M. Targeting of the SF/HGF receptor to the basolateral domain of polarized epithelial cells. J. Cell Biol. 1994, 125, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Weidner, K.M.; Vigna, E.; Gaudino, G.; Bardelli, A.; Ponzetto, C.; Narsimhan, R.P.; Hartmann, G.; Zarnegar, R.; Michalopoulos, G.K. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991, 10, 2867–2878. [Google Scholar] [CrossRef]
- Gentile, A.; Trusolino, L.; Comoglio, P.M. The Met tyrosine kinase receptor in development and cancer. Cancer Met. Rev. 2008, 27, 85–94. [Google Scholar] [CrossRef]
- Garajova, I.; Giovannetti, E.; Biasco, G.; Peters, G.J. c-Met. as a target for personalized therapy. Transl. Oncogenom. 2015, 7, 13–31. [Google Scholar] [CrossRef]
- Jeffers, M.; Rong, S.; Vande Woude, G.F. Hepatocyte growth factor/scatter factor-Met signaling in tumorigenicity and invasion/metastasis. J. Mol. Med. 1996, 74, 505–513. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar] [CrossRef]
- Salgia, R.; Sattler, M.; Scheele, J.; Stroh, C.; Felip, E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat. Rev. 2020, 87, 102022. [Google Scholar] [CrossRef] [PubMed]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Peschard, P.; Ishiyama, N.; Lin, T.; Lipkowitz, S.; Park, M. A conserved DpYR motif in the juxtamembrane domain of the Met receptor family forms an atypical c-Cbl/Cbl-b tyrosine kinase binding domain binding site required for suppression of oncogenic activation. J. Biol. Chem. 2004, 279, 29565–29571. [Google Scholar] [CrossRef]
- Taher, T.E.; Tjin, E.P.; Beuling, E.A.; Borst, J.; Spaargaren, M.; Pals, S.T. c-Cbl is involved in Met signaling in B cells and mediates hepatocyte growth factor-induced receptor ubiquitination. J. Immunol. 2002, 169, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar]
- Tan, Y.C.; Mirzapoiazova, T.; Won, B.M.; Zhu, L.; Srivastava, M.K.; Vokes, E.E.; Husain, A.N.; Batra, S.K.; Sharma, S.; Salgia, R. Differential responsiveness of MET inhibition in non-small-cell lung cancer with altered CBL. Sci. Rep. 2017, 7, 9192. [Google Scholar] [CrossRef]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef]
- Lee, J.; Kim, B.; Lee, S.B.; Jeong, Y.; Oh, Y.M.; Song, Y.J.; Jung, S.; Choi, J.; Lee, S.; Cheong, K.H.; et al. Cbl-independent degradation of Met: Ways to avoid agonism of bivalent Met-targeting antibody. Oncogene 2014, 33, 34–43. [Google Scholar] [CrossRef]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of Met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef]
- Benedettini, E.; Sholl, L.M.; Peyton, M.; Reilly, J.; Ware, C.; Davis, L.; Vena, N.; Bailey, D.; Yeap, B.Y.; Fiorentino, M.; et al. Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis. Am. J. Pathol. 2010, 177, 415–423. [Google Scholar] [CrossRef]
- Hong, L.; Zhang, J.; Heymach, J.V.; Le, X. Current and future treatment options for MET exon 14 skipping alterations in non-small cell lung cancer. Ther. Adv. Med. Oncol. 2021, 13, 1758835921992976. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.; Chung, L.Y.; Chau, S.L.; Lung, R.W.; Tong, C.Y.; Chow, C.; Tin, E.; Yu, Y.H.; et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Van De Kerkhove, C.; Wauters, E.; Van Mol, P. Capmatinib for the treatment of non-small cell lung cancer. Expert Rev. Anticancer Ther. 2019, 19, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Digumarthy, S.R.; Mendoza, D.P.; Zhang, E.W.; Lennerz, J.K.; Heist, R.S. Clinicopathologic and imaging features of non-small-cell lung cancer with MET exon 14 skipping mutations. Cancers 2019, 11, 2033. [Google Scholar] [CrossRef]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef]
- Fujino, T.; Suda, K.; Mitsudomi, T. Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin. Emerg. Drugs 2020, 25, 229–249. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Janne, P.A.; Skokan, M.; Finocchiaro, G.; Rossi, E.; Ligorio, C.; Zucali, P.A.; Terracciano, L.; Toschi, L.; Roncalli, M.; et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann. Oncol. 2009, 20, 298–304. [Google Scholar] [CrossRef]
- Shibata, T.; Uryu, S.; Kokubu, A.; Hosoda, F.; Ohki, M.; Sakiyama, T.; Matsuno, Y.; Tsuchiya, R.; Kanai, Y.; Kondo, T.; et al. Genetic classification of lung adenocarcinoma based on array-based comparative genomic hybridization analysis: Its association with clinicopathologic features. Clin. Cancer Res. 2005, 11, 6177–6185. [Google Scholar] [CrossRef][Green Version]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Zhao, X.; Weir, B.A.; LaFramboise, T.; Lin, M.; Beroukhim, R.; Garraway, L.; Beheshti, J.; Lee, J.C.; Naoki, K.; Richards, W.G.; et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005, 65, 5561–5570. [Google Scholar] [CrossRef] [PubMed]
- Calles, A.; Kwiatkowski, N.; Cammarata, B.K.; Ercan, D.; Gray, N.S.; Jänne, P.A. Tivantinib (ARQ 197) efficacy is independent of MET inhibition in non-small-cell lung cancer cell lines. Mol. Oncol. 2015, 9, 260–269. [Google Scholar] [CrossRef]
- Dimou, A.; Non, L.; Chae, Y.K.; Tester, W.J.; Syrigos, K.N. MET gene copy number predicts worse overall survival in patients with nonsmall cell lung cancer (NSCLC); a systematic review and meta-analysis. PLoS ONE 2014, 9, e107677. [Google Scholar] [CrossRef]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV non–small-cell lung cancer: METLung. J. Clin. Oncol. 2006, 35, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Shuster, D.; Orlov, S.; von Pawel, J.; Shepherd, F.A.; Ross, J.S.; Wang, Q.; Schwartz, B.; Akerley, W. Tivantinib in combination with erlotinib versus erlotinib alone for EGFR-mutant NSCLC: An exploratory analysis of the phase 3 MARQUEE study. J. Thorac. Oncol. 2018, 6, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.; von Pawel, J.; Novello, S.; Ramlau, R.; Favaretto, A.; Barlesi, F.; Akerley, W.; Orlov, S.; Santoro, A.; Spigel, D.; et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J. Clin. Oncol. 2015, 33, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Yu, H.; Ahn, M.-J.; Kim, S.; Cho, B.C.; Sequist, L.; Orlov, S.; Ottesen, L.H.; Verheijen, R.B.; Mellemgaard, A.; Wessen, J.; et al. TATTON phase Ib expansion cohort: Osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-amplified NSCLC after progression on prior first/second-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Cancer Res. 2019, 79, CT032. [Google Scholar] [CrossRef]
- Sequist, L.V.; Lee, J.S.; Han, J.Y.; Su, W.C.; Yang, J.C.; Yu, H.; Ottesen, L.H.; Verheijen, R.B.; Mellemgaard, A.; Wessen, J.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Bauml, J.; Cho, B.C.; Park, K.; Lee, K.H.; Cho, E.K.; Kim, D.W.; Kim, S.-W.; Haura, E.B.; Sabari, J.K.; Sanborn, R.E.; et al. Amivantamab in combination with lazertinib for the treatment of osimertinib-relapsed, chemotherapy-naïve EGFR mutant (EGFRm) non-small cell lung cancer (NSCLC) and potential biomarkers for response. J. Clin. Oncol. 2021, 39, 9006. [Google Scholar] [CrossRef]
- Okamoto, W.; Okamoto, I.; Tanaka, K.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Yamaguchi, H.; Arao, T.; Nishio, K.; Fukuoka, M.; et al. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol. Cancer Ther. 2010, 9, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Doshi, S.; Zhu, M. Pharmacokinetics and pharmacodynamics of rilotumumab: A decade of experience in preclinical and clinical cancer research. Br. J. Clin. Pharmacol. 2015, 80, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Rafique, I.; Floros, T.; Tran, P.; Gooding, W.E.; Villaruz, L.C.; Burns, T.F.; Friedland, D.M.; Petro, D.P.; Farooqui, M.; et al. Phase 1/2 study of rilotumumab (AMG 102), a hepatocyte growth factor inhibitor, and erlotinib in patients with advanced non-small cell lung cancer. Cancer 2017, 123, 2936–2944. [Google Scholar] [CrossRef]
- Tan, E.H.; Lim, W.T.; Ahn, M.J.; Ng, Q.S.; Ahn, J.S.; Shao-Weng, D.T.; Sun, J.M.; Han, M.; Payumo, F.C.; McKee, K.; et al. Phase 1b trial of ficlatuzumab, a humanized hepatocyte growth factor inhibitory monoclonal antibody, in combination with gefitinib in Asian patients with NSCLC. Clin. Pharmacol. Drug Dev. 2018, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ou, S.H.; Weiss, J.; Ades, S.; Conte, U.; Tang, Y.; Wang, S.; Murphy, D.; et al. Crizotinib in patients (pts) with MET-amplified non-small cell lung cancer (NSCLC): Updated safety and efficacy findings from a phase 1 trial. J. Clin. Oncol. 2018, 36, 9062. [Google Scholar] [CrossRef]
- Moro-Sibilot, D.; Cozic, N.; Pérol, M.; Mazières, J.; Otto, J.; Souquet, P.J.; Bahleda, R.; Wislez, M.; Zalcman, G.; Guibert, S.D.; et al. Crizotinib in c-MET- or ROS1-positive NSCLC: Results of the AcSé phase II trial. Ann. Oncol. 2019, 30, 1985–1991. [Google Scholar] [CrossRef]
- Strickler, J.H.; Weekes, C.D.; Nemunaitis, J.; Ramanathan, R.K.; Heist, R.S.; Morgensztern, D.; Angevin, E.; Bauer, T.M.; Yue, H.; Motwani, M.; et al. First-in-human phase I, dose-escalation and -expansion study of telisotuzumab vedotin, an antibody-drug conjugate targeting c-Met, in patients with advanced solid tumors. J. Clin. Oncol. 2019, 36, 3298–3306. [Google Scholar] [CrossRef]
- Award, M.M.; Leonardi, G.C.; Kravets, S.; Dahlberg, S.E.; Drilon, A.E.; Noonan, S.; Camidge, D.R.; Ou, S.-H.I.; Costa, D.B.; Gadgeel, S.M.; et al. Impact of MET inhibitors on survival among patients (pts) with MET exon 14 mutant (METdel14) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35, 8511. [Google Scholar] [CrossRef]
- Schuler, M.; Berardi, R.; Lim, W.T.; de Jonge, M.; Bauer, T.M.; Azaro, A.; Gottfried, M.; Han, J.Y.; Lee, D.H.; Wollner, M.; et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: Clinical and biomarker results from a phase I trial. Ann. Oncol. 2020, 31, 789–797. [Google Scholar] [CrossRef]
- Heist, R.S.; Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; Jonge, M.D.; Orlov, S.V.; et al. Capmatinib (INC280) in METΔEX14-mutated advanced NSCLC: Efficacy data from the phase 2 geometry MONO-1 study. J. Thorac. Oncol. 2019, 14, 1126. [Google Scholar] [CrossRef]
- Falchook, G.S.; Kurzrock, R.; Amin, H.M.; Xiong, W.; Fu, S.; Piha-Paul, S.A.; Janku, F.; Eskandari, G.; Catenacci, D.V.; Klevesath, M.; et al. First-in-man phase I trial of the selective MET inhibitor tepotinib in patients with advanced solid tumors. Clin. Cancer Res. 2020, 26, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Felip, E.; Dakai, H.; Patel, J.; Horn, L.; Veillon, R.; Griesinger, F.; Bruns, R.; Scheele, J.; Paik, P.J. Phase II Data for the MET Inhibitor Tepotinib in Patients with Advanced NSCLC and MET Exon 14-Skipping Mutations. J. Thorac. Oncol. 2018, 13, 347. [Google Scholar] [CrossRef]
- Klempner, S.J.; Borghei, A.; Hakimian, B.; Ali, S.M.; Ou, S.I. Intracranial activity of cabozantinibin MET exon 14-positive NSCLC with brain metastases. J. Thorac. Oncol. 2017, 12, 152–156. [Google Scholar] [CrossRef]
- Papaccio, F.; Della Corte, C.M.; Viscardi, G.; Di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the immune system: Relevance for cancer immunotherapy. Int. J. Mol. Sci. 2018, 19, 3595. [Google Scholar] [CrossRef] [PubMed]
- Kron, A.; Scheffler, M.; Heydt, C.; Ruge, L.; Schaepers, C.; Eisert, A.K.; Merkelbach-Bruse, S.; Riedel, R.; Nogova, L.; Fischer, R.N.; et al. Genetic heterogeneity of MET-aberrant NSCLC and its impact on the outcome of immunotherapy. J. Thorac. Oncol. 2021, 16, 572–582. [Google Scholar] [CrossRef]
- Sun, Z.J.; Wu, Y.; Hou, W.H.; Wang, Y.; Yuan, Q.Y.; Wang, H.J.; Yu, M. A novel bispecific c-MET/PD-1 antibody with therapeutic potential in solid cancer. Oncotarget 2017, 8, 29067–29079. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials. Available online: http://www.clinicaltrials.gov (accessed on 14 October 2021).


{kind=link}
| Detection Method | Technique | Detected MET Disorder | Material for Testing | Advantages | Disadvantages |
|---|---|---|---|---|---|
| NGS | Identification of the nucleotide sequence in the targeted regions/genes including MET (targeted sequencing); identification of substitutions, insertions/deletions, CNV, and rearrangements/fusions in a one test (CGP) | METex14 mutations, and other mutations, CNV, including amplification, rearrangements/fusions | DNA and RNA isolated from FFPE, cfDNA | Sensitive method with the possibility of DNA and RNA evaluation together | High costs of reagents, low availability of sequencers in laboratories; long samples preparation procedure for essential sequencing; the need for time-consuming bioinformatics analysis |
| qRT-PCR | Identification of mRNA with MET skipping mutation using molecular probes used for qPCR reactions, with prior rewriting of RNA sequences into cDNA | METex14, MET overexpression on RNA level | RNA isolated from FFPE material | Relatively simple and cheap method | qRT-PCR testing technique have insufficient sensitivity and specificity, may not detect all of splicing mutations; risk of RNA degradation, which requires special attention during preparation |
| FISH | Molecular fluorochrome-labeled probes attaching to the DNA in the cancer nucleidetected in fluorescence microscopy | CNV including amplification | FFPE cut on a microtome and placed on microscope slides | Identification of CNV directly in the cancer nuclei | Unable to identify the MET skipping mutation, and point mutation; fluorescence microscope is required |
| IHC | Detection of MET protein expression—visualization of the antigen-antibody complex and enzyme reaction, which is then viewed under light microscopy | MET protein expression | FFPE cut on a microtome and placed on microscope slides | IHC is a widely used method in diagnostic laboratories, its availability is high | Assessment of protein expression only, without the possibility of assessing the occurrence of the MET skipping mutation or CNV, including the amplification of the MET gene or overexpression on RNA level |
| Clinical Trial Identifier | Treatment Method | Stage of NSCLC | Phase | Estimated Enrollment | Status | MET Protein and MET Gene Diagnostics Strategy |
|---|---|---|---|---|---|---|
| NCT01456325 (METlung) | Onartuzumab + erlotinib vs. erlotinib + placebo | IIIB or IV | III | 499 | Completed | MET expression tested by IHC |
| NCT01244191 (MARQUEE) | Tivantinib + erlotinib vs. erlotinib + placebo | IIIB or IV | III | 1048 | Terminated | MET expression tested by IHC and MET GCN (gene copy number) tested by FISH |
| NCT01887886 | Erlotinib + onartuzumab vs. erlotinib + placebo | IIIB or IV | III | 10 | Completed | MET expression tested by IHC |
| NCT02031744 | Erlotinib + placebo vs. erlotinib + onartuzumab | IIIB/IV | III | 530 | Completed | MET expression tested by IHC |
| NCT04427072 (GEOMETRY-III) | Capmatinib vs. docetaxel | IIIB/IIIC or IV | III | 90 | Recruiting | METex14 mutation tested by NGS |
| NCT04816214 (GEOMETRY-E) | Capmatinib + osimertinib vs. chemotherapy (pemetrexed + cisplatin/ carboplatin) | IIIB/IIIC | III | 245 | Not yet recruiting | MET amplification measured in circulating tumor DNA (ctDNA) by real-time technique |
| NCT04677595 (GeoMETry-C) | Capmatinib | IIIB/IIIC or IV | II | 35 | Not yet recruiting | METex14 mutation assessed in circulating tumor DNA (ctDNA) by NGS |
| NCT04398940 | TQ-B3139 | IV | II | 71 | Recruiting | Differecnt tests for MET gene abnormalities |
| NCT03693339 (STARTER_cMET) | Capmatinib | IV | II | 27 | Recruiting | METex14 mutations tested by NGS and RT-PCR |
| NCT02099058 | Telisotuzumab vedotin + osimertinib vs. telisotuzumab vedotin + nivolumab vs. monotherapy telisotuzumab vedotin vs. telisotuzumab vedotin + erlotinib | IV (advance solid tumors) | I | 225 | Recruiting | MET expression tested by IHC |
| NCT03539536 (2018-001772-38) | Telisotuzumab vedotin | IIIB/IV | II | 310 | Recruiting | MET expression tested by IHC |
| NCT03993873 (TPX-0022-01) | TPX-0022 | IV | I | 120 | Recruiting | Genetic MET alterations including METex14 mutations, amplification, fusion or activating kinase mutation determined by NGS, FISH, quantitative polymerase chain reaction (qPCR) |
| NCT01639508 (12-097) | Cabozantinib | IV | II | 68 | Recruiting | MET overexpression, MET amplication or mutatation determined with different techniques |
| NCT02864992 (VISION) | Tepotinib | IIIB/IV | II | 330 | Recruiting | METex14 mutations in plasma and/or tissue determiend by NGS |
| NCT04084717 (CROME/ WI235747) | Crizotinib | IV | II | 50 | Recruiting | MET activating mutation (including METex14) or MET amplification tested in plasma or tissue with different technique including NGS |
| NCT03940703 (2019-001538-33) | Tepotinib + osimertinib vs. tepotinib | IIIB/IV | II | 120 | Recruiting | MET amplification determined by FISH and blood-based NGS |
| NCT04292119 (19-629) | Lorlatinib + crizotinib vs. lorlatinib + binimetinib vs. lorlatinib + TNO155 | IIIB/IV | I/II | 96 | Recruiting | Lack of MET testing, detection of ALK and ROS1 rearrangement |
| NCT01610336 | Capmatinib + gefitinib | - | II | 161 | Completed | |
| NCT04139317 (CINC280I12201) | Capmatinib (INC280) vs. pembrolizumab | IIIB/IV | II | 96 | Recruiting | MET gene copy number tested by FISH or MET overexpression tested by IHC |
| NCT04323436 (CINC280J12201) | Capmatinib (INC280) + spartalizumab (PDR001) vs. capmatinib + placebo | IIIB/IV | II | 270 | Recruiting | METex14 mutations tested by NGS |
| NCT03333343 (CEGF816X2102) | EGF816 + INC280 | IIIB/IV | I | 157 | Recruiting | - |
| NCT04606771 | Osimertinib + savolitinib vs. savolitinib + placebo | IIIB/IV | II | 56 | Recruiting | MET amplification tested by FISH |
| NCT03778229 (SAVANNAH) | Osimertinib + savolitinib | IIIB/IV | II | 259 | Recruiting | MET amplifiecation and MET overexpresion tested by FISH or IHC |
| NCT03944772 (ORCHARD) | Osimertinib + savolitinib vs. osimertinib + gefitinib vs. osimertinib + necitumumab vs. carboplatin + pemetrexed + durvalumab vs. observational cohort—no study drug vs. osimertinib + alectinib vs. osimertinib + selpercatinib | IIIB/IV | II | 150 | Recruiting | - |
| NCT02954991 | Glesatinib + nivolumab vs. sitravatinib + nivolumab vs. mocetinostat + nivolumab | IIIB/IV | II | 206 | Active, not recruiting | - |
| NCT03906071 (SAPPHIRE) | Nivolumab + sitravatinib vs. docetaxel | IV | III | 532 | Recruiting | Testing for EGFR mutations, ROS1 fusions, ALK mutations or ALK fusions, MET not tested |
| NCT02664935 (ISRCTN38344105) | AZD4547 vs. vistusertib vs. palbociclib vs. crizotinib vs. selumetinib + docetaxel vs. AZD5363 vs. osimertinib vs. durvalumab vs. sitravatinib + AZD6738 | III/IV | II | 549 | Recruiting | - |
| NCT04739358 | Tepotinib | IV | I/II | 65 | Not yet recruiting | METex14 mutations tested by NGS, MET amplifications tested by FISH, MET fusions tested by NGS |
| NCT04131543 (CRETA) | Cabozantinib | IIIB/IV | II | 25 | Recruiting | RET rearrangement tested by FISH or NGS, MET not tested |
| NCT04173338 (IST-65) | Cabozantinib + pemetrexed | IIIB/IV | I | 30 | Recruiting | - |
| NCT04310007 (NCI-2020-01541) | Cabozantinib vs. cabozantinib + nivolumab vs. standard chemotherapy | III, IIIA, IIIB, IIIC, IVA, IV | II | 142 | Recruiting | METex14 mutations tested by NGS, MET amplification tested by FISH |
| NCT02795156 (SCRI PRO 10) | Afatinib vs. regorafenib vs. cabozantinib | - | II | 160 | Recrutiung | - |
| NCT04514484 (NCI-2020-05956) | Cabozantinib + nivolumab | IV | I | 18 | Recrutiung | - |
| NCT03170960 (XL184-021) | Cabozantinib + atezolizumab | IV | I/II | 1732 | Recruiting | - |
| NCT04148066 (TATIN) | Osimertinib + crizotinib | IV | - | 30 | Recruiting | - |
| NCT02034981 (AcSé) | Crizotinib | IV | II | 246 | Active, not recruiting | One proven specific alterations among ALK, MET, RON, and ROS1 genes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terlecka, P.; Krawczyk, P.; Grenda, A.; Milanowski, J. MET Gene Dysregulation as a Promising Therapeutic Target in Lung Cancer—A Review. J. Pers. Med. 2021, 11, 1370. https://doi.org/10.3390/jpm11121370
Terlecka P, Krawczyk P, Grenda A, Milanowski J. MET Gene Dysregulation as a Promising Therapeutic Target in Lung Cancer—A Review. Journal of Personalized Medicine. 2021; 11(12):1370. https://doi.org/10.3390/jpm11121370
Chicago/Turabian StyleTerlecka, Paulina, Paweł Krawczyk, Anna Grenda, and Janusz Milanowski. 2021. "MET Gene Dysregulation as a Promising Therapeutic Target in Lung Cancer—A Review" Journal of Personalized Medicine 11, no. 12: 1370. https://doi.org/10.3390/jpm11121370
APA StyleTerlecka, P., Krawczyk, P., Grenda, A., & Milanowski, J. (2021). MET Gene Dysregulation as a Promising Therapeutic Target in Lung Cancer—A Review. Journal of Personalized Medicine, 11(12), 1370. https://doi.org/10.3390/jpm11121370