
Analysis of Rare Variants in Genes Related to Lipid Metabolism in Patients with Familial Hypercholesterolemia in Western Siberia (Russia)

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Isolation of Genomic DNA
2.3. Genome Library Preparation, Sequencing, and Bioinformatic Analysis
- −
- Big population frequency databases, such as gnomAD (available online: https://gnomad.broadinstitute.org/ [accessed on 4 October 2021]), with some help of databases on specific populations, like Greater Middle East (GME) Variome Project (available online: http://igm.ucsd.edu/gme/ [accessed on 4 October 2021]), AbraOM (Brazilian genomic variants) (available online: https://abraom.ib.usp.br/ [accessed on 4 October 2021]), and Korean Personal Genome Project (available online: http://opengenome.net/Main_Page [accessed on 4 October 2021]).
- −
- Databases representing in silico prediction tools, such as dbNSFP (available online: https://sites.google.com/site/jpopgen/dbNSFP [accessed on 4 October 2021]), which contains data from more than 30 pathogenicity prediction tools (e.g., MutationTaster2, SIFT, PROVEAN, and Polyphen2), and from 10 conservation prediction tools (e.g., phastCons, GERP++, and SiPhy). For pathogenicity prediction tools, we set thresholds according to respective authors’ recommendations; additionally, for conservation prediction tools, we used one common threshold, 0.7; therefore, we assumed a variant to be conserved if its conservation score was greater than the scores of ≥70% other variants. Additionally, we used databases dbscSNV and regSNP-intron for variants that may have an effect on splicing. Nevertheless, all these databases were only a supplementary tool, and they contributed little to the summary measure of pathogenicity.
- −
- Gene-based phenotype databases (e.g., OMIM).
- −
- Clinical significance databases: ClinVar (available online: https://www.ncbi.nlm.nih.gov/clinvar/ [accessed on 4 October 2021]), Human Gene Mutation Database (HGMD) (available online: http://www.hgmd.cf.ac.uk/ [accessed on 4 October 2021]), and Leiden Open Variation Database (available online: https://www.lovd.nl/ [accessed on 4 October 2021]); variants described as benign (B) or likely benign (LB) were also excluded from further analysis.
- −
- PubMed (available online: https://pubmed.ncbi.nlm.nih.gov/ [accessed on 4 October 2021]) and some other article databases as a source of information on specific clinical cases.
2.4. Verification of Findings
2.5. Multiplex Ligation-Dependent Probe Amplification (MLPA)
3. Results
3.1. LDLR
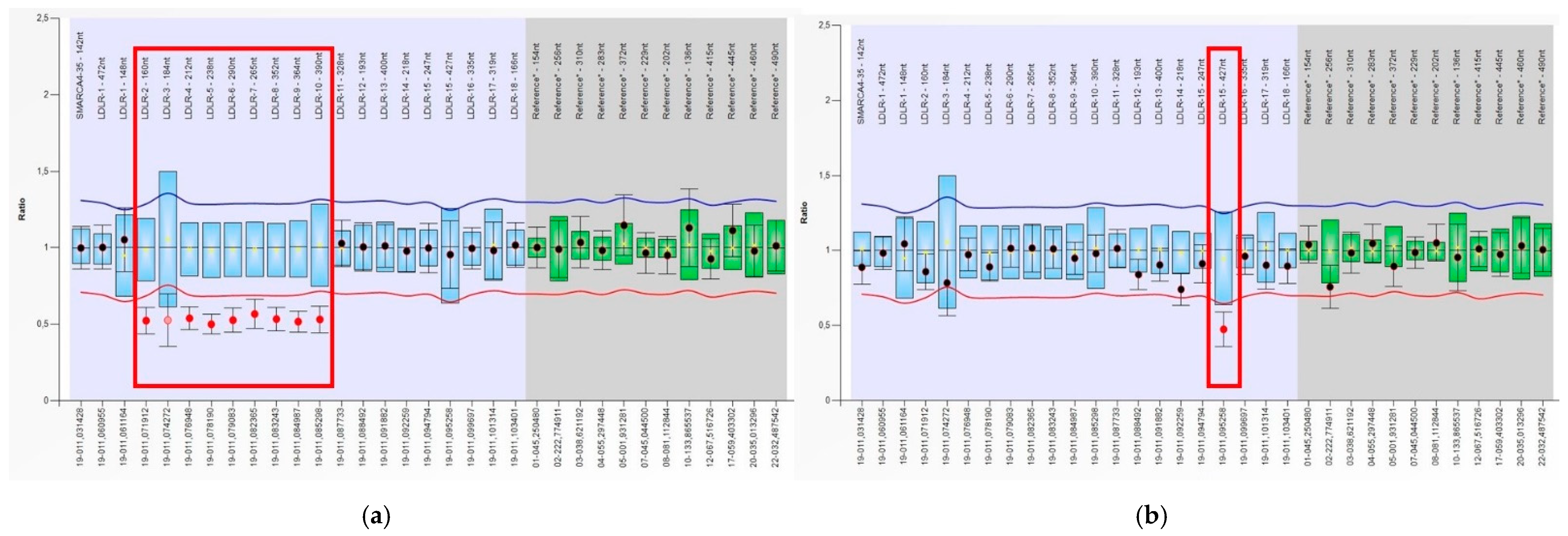
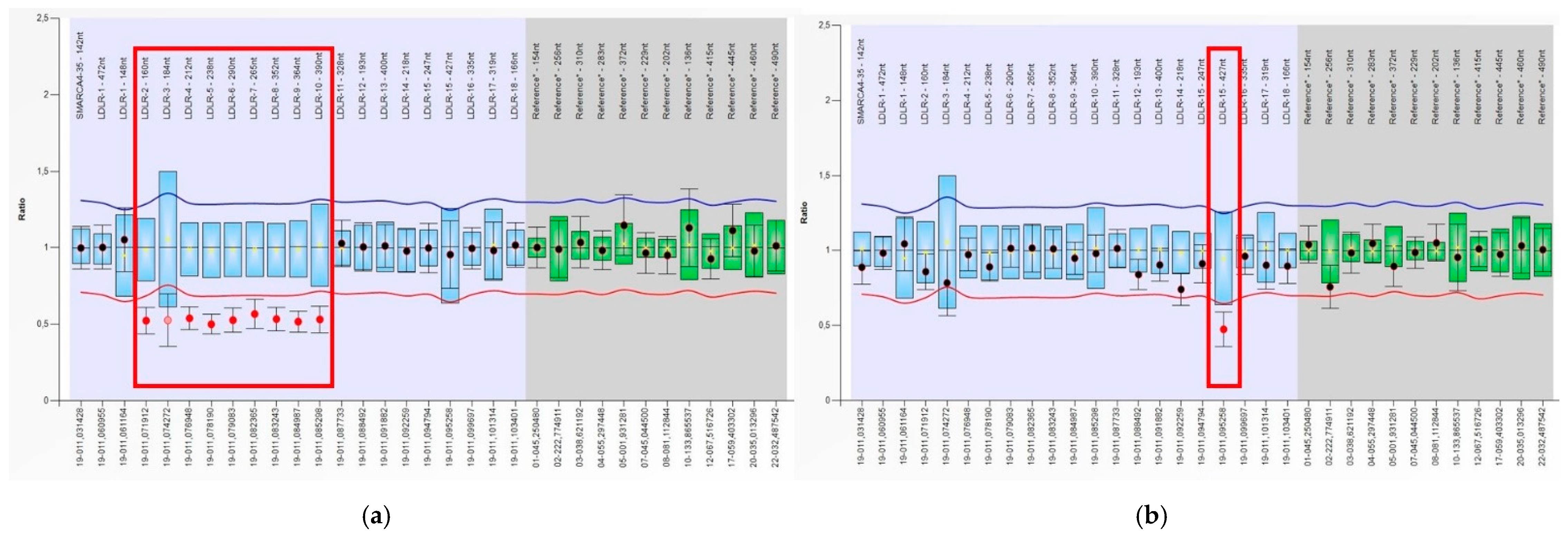
3.2. Identification of Deletions and Duplications in the LDLR Gene by MLPA
3.3. APOB
3.4. ABCG5
3.5. APOC3
3.6. LPL
3.7. SREBF1
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ezhov, M.V.; Bazhan, S.S.; Ershova, A.I.; Meshkov, A.N.; Sokolov, A.A.; Kukharchuk, V.V.; Gurevich, V.S.; Voevoda, M.I.; Sergienko, I.V.; Shakhtshneider, E.V.; et al. Clinical guidelines for familial hypercholesterolemia. Ateroskleroz 2019, 15, 58–98. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjouke, B.; Kusters, D.M.; Kindt, I.; Besseling, J.; Defesche, J.C.; Sijbrands, E.J.; Roeters van Lennep, J.E.; Stalenhoef, A.F.; Wiegman, A.; de Graaf, J.; et al. Homozygous autosomal dominant hypercholesterolemia in the Netherlands: Prevalence, genotype–phenotype relationship, and clinical outcome. Eur. Heart J. 2015, 36, 560–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Santos, R.D.; Gidding, S.S.; Hegele, R.A.; Cuchel, M.A.; Barter, P.J.; Watts, G.F.; Baum, S.J.; Catapano, A.L.; Chapman, M.J.; Defesche, J.C.; et al. International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Defining severe familial hypercholesterolaemia and the implications for clinical management: A consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016, 4, 850–861. [Google Scholar] [CrossRef]
- Wiegman, A. Lipid Screening, Action, and Follow-up in Children and Adolescents. Curr Cardiol. Rep. 2018, 20, 80. [Google Scholar] [CrossRef]
- Wiegman, A.; Gidding, S.S.; Watts, G.F.; Chapman, M.J.; Ginsberg, H.N.; Cuchel, M.; Ose, L.; Averna, M.; Boileau, C.; Borén, J.; et al. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia in children and adolescents: Gaining decades of life by optimizing detection and treatment. Eur. Heart J. 2015, 36, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Brautbar, A.; Leary, E.; Rasmussen, K.; Wilson, D.P.; Steiner, R.D.; Virani, S. Genetics of familial hypercholesterolemia. Curr. Atheroscler. Rep. 2015, 17, 491. [Google Scholar] [CrossRef]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Convened by the Familial Hypercholesterolemia Foundation. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Motazacker, M.M.; Pirruccello, J.; Huijgen, R.; Do, R.; Gabriel, S.; Peter, J.; Kuivenhoven, J.A.; Defesche, J.C.; Kastelein, J.J.; Hovingh, G.K.; et al. Advances in genetics show the need for extending screening strategies for autosomal dominant hypercholesterolaemia. Eur. Heart J. 2012, 33, 1360–1366. [Google Scholar] [CrossRef] [Green Version]
- van der Graaf, A.; Avis, H.J.; Kusters, D.M.; Vissers, M.N.; Hutten, B.A.; Defesche, J.C.; Huijgen, R.; Fouchier, S.W.; Wijburg, F.A.; Kastelein, J.J.; et al. Molecular basis of autosomal dominant hypercholesterolemia: Assessment in a large cohort of hypercholesterolemic children. Circulation 2011, 123, 1167–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Taranto, M.D.; Giacobbe, C.; Fortunato, G. Familial hypercholesterolemia: A complex genetic disease with variable phenotypes. Eur J. Med. Genet. 2020, 63, 103831. [Google Scholar] [CrossRef]
- Berberich, A.J.; Hegele, R.A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2019, 16, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Manickam, K.; Jones, L.K.; Wright, E.A.; Hartzel, D.N.; Gonzaga-Jauregui, C.; O′Dushlaine, C.; Leader, J.B.; Lester Kirchner, H.; Lindbuchler, D.M.; et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016, 354, aaf7000. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.J.; Peloso, G.M.; Yu, H.; Butterworth, A.S.; Wang, X.; Mahajan, A.; Saleheen, D.; Emdin, C.; Alam, D.; Alves, A.C.; et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet. 2017, 49, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Peloso, G.M.; Auer, P.L.; Bis, J.C.; Voorman, A.; Morrison, A.C.; Stitziel, N.O.; Brody, J.A.; Khetarpal, S.A.; Crosby, J.R.; Fornage, M.; et al. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am. J. Hum. Genet. 2014, 94, 223–232. [Google Scholar] [CrossRef] [Green Version]
- van der Laan, S.W.; Harshfield, E.L.; Hemerich, D.; Stacey, D.; Wood, A.M.; Asselbergs, F.W. From lipid locus to drug target through human genomics. Cardiovasc. Res. 2018, 114, 1258–1270. [Google Scholar] [CrossRef]
- Leren, T.P.; Finborud, T.H.; Manshaus, T.E.; Ose, L.; Berge, K.E. Diagnosis of familial hypercholesterolemia in general practice using clinical diagnostic criteria or genetic testing as part of cascade genetic screening. Community Genet. 2008, 11, 26–35. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol: Chloroform. CSH Protoc. 2006, 2006, pdb.prot4455. [Google Scholar] [CrossRef]
- Fouchier, S.W.; Dallinga-Thie, G.M.; Meijers, J.C.; Zelcer, N.; Kastelein, J.J.; Defesche, J.C.; Hovingh, G.K. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 2014, 115, 552–555. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.X.; Wu, N.Q.; Sun, D.; Liu, H.H.; Jin, J.L.; Li, S.; Guo, Y.L.; Zhu, C.G.; Gao, Y.; Dong, Q.T.; et al. Application of expanded genetic analysis in the diagnosis of familial hypercholesterolemia in patients with very early-onset coronary artery disease. J. Transl. Med. 2018, 16, 345. [Google Scholar] [CrossRef] [Green Version]
- Fouchier, S.W.; Defesche, J.C. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr. Opin. Lipidol. 2013, 24, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Vinje, T.; Wierød, L.; Leren, T.P.; Strøm, T.B. Prevalence of cholesteryl ester storage disease among hypercholesterolemic subjects and functional characterization of mutations in the lysosomal acid lipase gene. Mol. Genet. Metab. 2018, 123, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Lamiquiz-Moneo, I.; Baila-Rueda, L.; Bea, A.M.; Mateo-Gallego, R.; Pérez-Calahorra, S.; Marco-Benedí, V.; Martín-Navarro, A.; Ros, E.; Cofán, M.; Rodríguez-Rey, J.C.; et al. ABCG5/G8 gene is associated with hypercholesterolemias without mutation in candidate genes and noncholesterol sterols. J. Clin. Lipidol. 2017, 11, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Zakharova, F.M.; Damgaard, D.; Mandelshtam, M.Y.; Golubkov, V.I.; Nissen, P.H.; Nilsen, G.G.; Stenderup, A.; Lipovetsky, B.M.; Konstantinov, V.O.; Denisenko, A.D.; et al. Familial hypercholesterolemia in St-Petersburg: The known and novel mutations found in the low-density lipoprotein receptor gene in Russia. BMC Med. Genet. 2005, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakhtshneider, E.; Orlov, P.; Ivanoshchuk, D.; Makarenkova, K.; Ragino, Y.; Voevoda, M. Analysis of the LDLR gene variability in patients with familial hypercholesterolemia in Russia using targeted high throughput resequencing. Atherosclerosis 2017, 263, e227. [Google Scholar] [CrossRef]
- Shakhtshneider, E.; Ivanoshchuk, D.; Orlov, P.; Timoshchenko, O.; Voevoda, M. Analysis of the LDLR, APOB, PCSK9 and LDLRAP1 genes variability in patients with familial hypercholesterolemia in West Siberia using targeted high throughput resequencing. Atherosclerosis 2019, 287, e285. [Google Scholar] [CrossRef]
- Vasilyev, V.; Zakharova, F.; Bogoslovskay, T.; Mandelshtam, M. Familial Hypercholesterolemia in Russia: Three Decades of Genetic Studies. Front. Genet. 2020, 11, 550591. [Google Scholar] [CrossRef]
- Miroshnikova, V.V.; Romanova, O.V.; Ivanova, O.N.; Fedyakov, M.A.; Panteleeva, A.A.; Barbitoff, Y.A.; Muzalevskaya, M.V.; Urazgildeeva, S.A.; Gurevich, V.S.; Urazov, S.P.; et al. Identification of novel variants in the LDLR gene in Russian patients with familial hypercholesterolemia using targeted sequencing. Biomed. Rep. 2021, 14, 15. [Google Scholar] [CrossRef]
- Semenova, A.E.; Sergienko, I.V.; García-Giustiniani, D.; Monserrat, L.; Popova, A.B.; Nozadze, D.N.; Ezhov, M.V. Verification of underlying genetic cause in a cohort of Russian patients with familial hypercholesterolemia using targeted next generation sequencing. J. Cardiovasc. Dev. Dis. 2020, 7, 16. [Google Scholar] [CrossRef]
- Meshkov, A.N.; Malyshev, P.P.; Kukharchuk, V.V. Familial hypercholesterolemia in Russia: Genetic and phenotypic characteristics. Ter. Arkh. 2009, 81, 23–28. [Google Scholar] [PubMed]
- Voevoda, M.; Shakhtshneider, E.; Orlov, P.; Ivanoshchuk, D.; Ivanova, M.; Nikitin, Y.; Malyutina, S. The mutation R3500Q of apolipoprotein B in caucasian population of west Siberia and in patients with highest total cholesterol level. Atherosclerosis 2014, 235, e100. [Google Scholar] [CrossRef]
- Pamplona-Cunha, H.; Medeiros, M.F.; Sincero, T.C.M.; Back, I.C.; Silva, E.L.D. Compound Heterozygous Familial Hypercholesterolemia Caused by LDLR Variants. Arq. Bras. Cardiol. 2020, 115, 587–589. [Google Scholar] [CrossRef]
- Shakhtshneider, E.V.; Ivanoshchuk, D.E.; Makarenkova, K.V.; Orlov, P.S.; Timoshchenko, O.V.; Bazhan, S.S.; Nikitin, Y.P.; Voevoda, M.I. Cascade genetic screening in diagnostics of heterozygous familial hypercholesterolemia: Clinical case. Russ. J. Cardiol. 2017, 6, 178–179. [Google Scholar] [CrossRef] [Green Version]
- Shakhtshneider, E.; Ivanoshchuk, D.; Makarenkova, K.; Orlov, P.; Timoshchenko, O.; Bazhan, S.; Voevoda, M. Clinical case: The development of heterozygous familial hypercholesterolemia in a patient with rs879255191 in LDLR gene. Atherosclerosis 2018, 275, e179. [Google Scholar] [CrossRef]
- Marduel, M.; Carrié, A.; Sassolas, A.; Devillers, M.; Carreau, V.; Di Filippo, M.; Erlich, D.; Abifadel, M.; Marques-Pinheiro, A.; Munnich, A.; et al. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Hum. Mutat. 2010, 31, E1811–E1824. [Google Scholar] [CrossRef] [Green Version]
- Koivisto, P.V.; Koivisto, U.M.; Miettinen, T.A.; Kontula, K. Diagnosis of heterozygous familial hypercholesterolemia. DNA analysis complements clinical examination and analysis of serum lipid levels. Arterioscler. Thromb. 1992, 12, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/17890/ (accessed on 1 October 2021).
- Available online: https://gnomad.broadinstitute.org/variant/2–44059195-G-C?dataseT=gnomad_r2_1 (accessed on 1 October 2021).
- Helgadottir, A.; Thorleifsson, G.; Alexandersson, K.F.; Tragante, V.; Thorsteinsdottir, M.; Eiriksson, F.F.; Gretarsdottir, S.; Björnsson, E.; Magnusson, O.; Sveinbjornsson, G.; et al. Genetic variability in the absorption of dietary sterols affects the risk of coronary artery disease. Eur. Heart J. 2020, 41, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, A.B.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjorg-Hansen, A.; Tybjorg-Hansen, A. Loss-of-Function Mutations in APOC3 and Risk of Ischemic Vascular Disease. N. Engl. J. Med. 2014, 371, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surendran, R.P.; Visser, M.E.; Heemelaar, S.; Wang, J.; Peter, J.; Defesche, J.C.; Kuivenhoven, J.A.; Hosseini, M.; Péterfy, M.; Kastelein, J.J.; et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J. Intern. Med. 2012, 272, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Liu, M.S.; Chitayat, D.; Bruin, T.; Beisiegel, U.; Benlian, P.; Foubert, L.; De Gennes, J.L.; Funke, H.; Forsythe, I.; et al. Recurrent missense mutations at the first and second base of codon Arg243 in human lipoprotein lipase in patients of different ancestries. Hum. Mutat. 1994, 3, 52–58. [Google Scholar] [CrossRef]
- Neuner, J.; Dimmock, D.; Kirschner, A.P.; Beaudry, H.; Paradowski, J.; Orlando, L. Results and Lessons of a Pilot Study of Cascade Screening for Familial Hypercholesterolemia in US Primary Care Practices. J. Gen. Intern. Med. 2020, 35, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, V.; Banescu, C.; Dobreanu, M. Molecular diagnosis methods in familial hypercholesterolemia. Anatol. J. Cardiol. 2020, 23, 120–127. [Google Scholar] [CrossRef]
- McGowan, M.P.; Cuchel, M.; Ahmed, C.D.; Khera, A.; Weintraub, W.S.; Wilemon, K.A.; Ahmad, Z. A proof-of-concept study of cascade screening for Familial Hypercholesterolemia in the US, adapted from the Dutch model. Am. J. Prev. Cardiol. 2021, 11, 100170. [Google Scholar] [CrossRef]
- Peterson, A.L.; Bang, M.; Block, R.C.; Wong, N.D.; Karalis, D.G. Cascade Screening and Treatment Initiation in Young Adults with Heterozygous Familial Hypercholesterolemia. J. Clin. Med. 2021, 10, 3090. [Google Scholar] [CrossRef]
- Truong, T.H.; Do, D.L.; Kim, N.T.; Nguyen, M.T.; Le, T.T.; Le, H.A. Genetics, Screening, and Treatment of Familial Hypercholesterolemia: Experience Gained From the Implementation of the Vietnam Familial Hypercholesterolemia Registry. Front. Genet. 2020, 14, 914. [Google Scholar] [CrossRef]
- Garrahy, E.; Heal, C.; Hespe, C.M.; Radford, J.; Watts, G.F.; Brett, T. Familial hypercholesterolaemia and cascade testing in general practice: Lessons from COVID-19. Aust J. Gen. Pract. 2020, 49, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.P.; Reeskamp, L.F.; Hovingh, G.K. Familial Hypercholesterolemia: The Most Common Monogenic Disorder in Humans. J. Am. Coll. Cardiol. 2020, 26, 2567–2569. [Google Scholar] [CrossRef] [PubMed]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; Cooper, J.A.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of low density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013, 381, e1293–e1301. [Google Scholar] [CrossRef] [Green Version]
- Masana, L.; Ibarretxea, D.; Rodríguez-Borjabad, C.; Plana, N.; Valdivielso, P.; Pedro-Botet, J.; Civeira, F.; López-Miranda, J.; Guijarro, C.; Mostaza, J.; et al. Toward a new clinical classification of patients with familial hypercholesterolemia: One perspective from Spain. Atherosclerosis. 2019, 287, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K.; Baliga, R.R. Genetics of Dyslipidemia and Ischemic Heart Disease. Curr. Cardiol. Rep. 2017, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, A.M.; Alves, A.C.; Bourbon, M. Mutational analysis of a cohort with clinical diagnosis of familial hypercholesterolemia: Considerations for genetic diagnosis improvement. Genet. Med. 2016, 18, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Meshkov, A.; Ershova, A.; Kiseleva, A.; Zotova, E.; Sotnikova, E.; Petukhova, A.; Zharikova, A.; Malyshev, P.; Rozhkova, T.; Blokhina, A.; et al. The LDLR, APOB, and PCSK9 Variants of Index Patients with Familial Hypercholesterolemia in Russia. Genes 2021, 12, 66. [Google Scholar] [CrossRef]
- Averkova, A.O.; Brazhnik, V.A.; Speshilov, G.I.; Rogozhina, A.A.; Koroleva, O.S.; Zubova, E.A.; Koroleva, O.S.; Zubova, E.A.; Galyavich, A.S.; Tereshenko, S.N.; et al. Target sequencing in patients with clinically diagnosed hereditary lipid metabolism disorders and acute coronary syndrome. Bull. Russ. State Med. Univ. 2020, 5, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Vlad, C.-E.; Foia, L.G.; Popescu, R.; Popa, I.; Aanicai, R.; Reurean-Pintilei, D.; Toma, V.; Florea, L.; Kanbay, M.; Covic, A. Molecular Genetic Approach and Evaluation of Cardiovascular Events in Patients with Clinical Familial Hypercholesterolemia Phenotype from Romania. J. Clin. Med. 2021, 10, 1399. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| M ± SD * | Minimum | Maximum | |
|---|---|---|---|
| Glucose, mmol/L | 5.7 ± 1.2 | 4.0 | 10.4 |
| Total cholesterol, mmol/L (mg/dL) | 8.6 ± 3.4 (332.5 ± 131.4) | 3.4 (131.4) | 25.0 (966.5) |
| Triglycerides, mmol/L (mg/dL) | 1.8 ± 1.4 (157.5 ± 122.5) | 0.4 (35.0) | 17.4 (1522.5) |
| HDL-C, mmol/L (mg/dL) | 1.4 ± 0.4 (54.1 ± 15.5) | 0.54 (20.9) | 2.2 (85.1) |
| LDL-C, mmol/L (mg/dL) | 5.6 ± 2.3 (216.5 ± 88.9) | 1.11 (42.9) | 11.94 (461.6) |
| Body–mass index, m2/kg | 27.2 ± 4.6 | 19.0 | 39.0 |
| Age, years | 46.0 ± 13.9 | 20 | 73 |
| Patient ID | dbSNP ID | Position on Chromosome (GRCh38) | Nucleotide Substitution | Amino Acid Substitution | MAF According to Database GnomAD | Clinical Effect According to Database ClinVar | References for Russia |
|---|---|---|---|---|---|---|---|
| LDLR Gene | |||||||
| P28, P40, P41, P42, P55, P56 | rs121908038 | 19:11113293 | c.1202T > A | p.Leu401His | ND | Likely Pathogenic | Zakharova et al., 2005 [26]; Shakhtshneider et al., 2017 [27]; Shakhtshneider et al., 2019 [28]; Vasilyev et al., 2020 [29]; Miroshnikova et al., 2021 [30] |
| P45 | rs137853964 | 19:11129602 | c.2479G > A | p.Val827Ile | A = 0.001006 | Likely Pathogenic | Shakhtshneider et al., 2017 [27]; Shakhtshneider et al., 2019 [28]; Vasilyev et al., 2020 [29] |
| Р22, P36, P58 | rs28942078 | 19:11113376 | c.1285G > A | p.Val429Met | A = 0.000012 | Pathogenic/Likely Pathogenic | - |
| P65 | rs539080792 | 19:11221396 | c.1009G > A | p.Glu337Lys | A = 0.000104 | Uncertain Significance | Shakhtshneider et al., 2017 [27]; Shakhtshneider et al., 2019 [28]; Vasilyev et al., 2020 [29] |
| P47 | rs570942190 | 19:11113337 | c.1246C > T | p.Arg416Trp | T = 0.000024 | Not reported in ClinVar | Shakhtshneider et al., 2017 [27]; Shakhtshneider et al., 2019 [28]; Vasilyev et al., 2020 [29] |
| P67, P68 | rs755757866 | 19:11110730 | c.1019G > T | p.Cys340Tyr | T = 0.000008 | Likely Pathogenic | Shakhtshneider et al., 2017 [27]; Shakhtshneider et al., 2019 [28]; Vasilyev et al., 2020 [29] |
| Р7 | rs761954844 | 19:11110697 | c.986G > A | p.Cys329Tyr | A = 0.000016 | Likely Pathogenic | Zakharova et al., 2005 [26]; Shakhtshneider et al., 2019 [28]; Semenova et al., 2020 [31]; Vasilyev et al., 2020 [29]; Miroshnikova et al., 2021 [30] |
| P35 | rs879254566 | 19:11105440 | c.534TT > G | p.Asp178Glu | ND | Pathogenic/ Likely Pathogenic | Shakhtshneider et al., 2019 [28]; Vasilyev et al., 2020 [29] |
| P38, P39 | rs879254721 | 19:11107496 | c.922G > A | p.Glu308Lys | ND | Pathogenic | Semenova et al., 2020 [31]; Vasilyev et al., 2020 [30]; |
| P2 | rs879254980 | 19:11116179 | c.1672G > T | p.Glu558Ter | ND | Pathogenic | - |
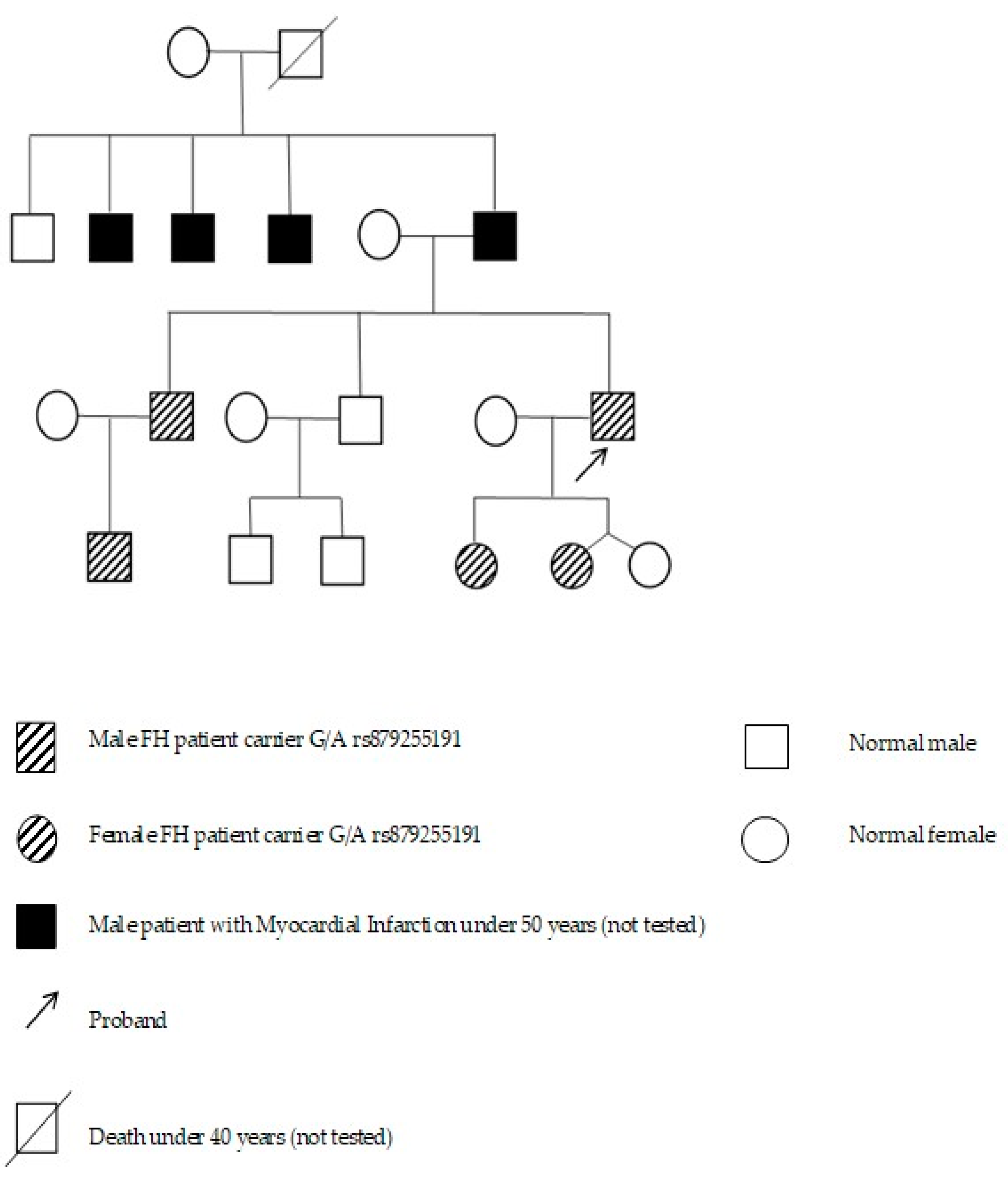
| P24, P25, P26, P81, P82 | rs879255191 | 19:11128090 | c.2389 + 5G > A | - | ND | Conflicting Interpretations of Pathogenicity | Meshkov et. Al. [32]; Shakhtshneider et al., 2019 [28]; Vasilyev et al., 2020 [29] |
| P52 | rs875989907 | 19:11106666 | c.796G > A | p.Asp266Asn | A = 0.000012 | Pathogenic | Shakhtshneider et al., 2017 [27] |
| rs879254769 | 19:11110765 | c.1054T > C | p.Cys352Ser | ND | Likely Pathogenic | Shakhtshneider et al., 2017 [27] | |
| P10 | rs875989894 | 19:11213415 | c.266G > C | p.Cys89Ser | ND | Pathogenic/Likely Pathogenic | - |
| ND | 19:11222252 | c.1123T > G | p.Tyr375Asp | ND | Novel variant | - | |
| APOB Gene | |||||||
| P11, P15, P71 | rs5742904 | 2:21006288 | c.10580G > A | p.Arg3527Gln | T = 0.000275 | Pathogenic | Voevoda et al. 2014 [33]; Shakhtshneider et al., 2019 [28]; Miroshnikova et al., 2021 [30] |
| ABCG5 Gene | |||||||
| P74 | rs145164937 | 2:43832056 | c.293C > G | p.Ala98Gly | C = 0.002223 | Conflicting interpretations of pathogenicity | - |
| APOC3 Gene | |||||||
| P59 | rs138326449 | 11:116830638 | c.55 + 1G > A | - | C = 0.002244 | Conflicting interpretations of pathogenicity | - |
| LPL Gene | |||||||
| P9 | rs118204077 | 8:19955873 | c.808C > T | p.Arg270Cys | C = 0.0001 | Pathogenic | - |
| SREBF1 Gene | |||||||
| P83, P84 | rs115855236 | 17:17820281 | c.422C > T | p.Pro141Leu | A = 0.001210 | Not reported in ClinVar | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakhtshneider, E.; Ivanoshchuk, D.; Timoshchenko, O.; Orlov, P.; Semaev, S.; Valeev, E.; Goonko, A.; Ladygina, N.; Voevoda, M. Analysis of Rare Variants in Genes Related to Lipid Metabolism in Patients with Familial Hypercholesterolemia in Western Siberia (Russia). J. Pers. Med. 2021, 11, 1232. https://doi.org/10.3390/jpm11111232
Shakhtshneider E, Ivanoshchuk D, Timoshchenko O, Orlov P, Semaev S, Valeev E, Goonko A, Ladygina N, Voevoda M. Analysis of Rare Variants in Genes Related to Lipid Metabolism in Patients with Familial Hypercholesterolemia in Western Siberia (Russia). Journal of Personalized Medicine. 2021; 11(11):1232. https://doi.org/10.3390/jpm11111232
Chicago/Turabian StyleShakhtshneider, Elena, Dinara Ivanoshchuk, Olga Timoshchenko, Pavel Orlov, Sergey Semaev, Emil Valeev, Andrew Goonko, Nataliya Ladygina, and Mikhail Voevoda. 2021. "Analysis of Rare Variants in Genes Related to Lipid Metabolism in Patients with Familial Hypercholesterolemia in Western Siberia (Russia)" Journal of Personalized Medicine 11, no. 11: 1232. https://doi.org/10.3390/jpm11111232
APA StyleShakhtshneider, E., Ivanoshchuk, D., Timoshchenko, O., Orlov, P., Semaev, S., Valeev, E., Goonko, A., Ladygina, N., & Voevoda, M. (2021). Analysis of Rare Variants in Genes Related to Lipid Metabolism in Patients with Familial Hypercholesterolemia in Western Siberia (Russia). Journal of Personalized Medicine, 11(11), 1232. https://doi.org/10.3390/jpm11111232