
Epigenotype, Genotype, and Phenotype Analysis of Taiwanese Patients with Silver–Russell Syndrome

 ,
, 
 , , , and
, , , and 
Abstract
1. Introduction
2. Patients and Methods
2.1. Study Population
2.2. Clinical Assessments
2.3. Molecular Studies
2.4. Data and Statistical Analysis
3. Results
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
Consent for Publication
Abbreviations
References
- Silver, H.K.; Kiyasu, W.; George, J.; Deamer, W.C. Syndrome of congenital hemihypertrophy, shortness of stature, and elevated urinary gonadotropins. Pediatrics 1953, 12, 368–376. [Google Scholar]
- Russell, A. A syndrome of intra-uterine dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples). J. R. Soc. Med. 1954, 47, 1040–1044. [Google Scholar]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef]
- Luk, H.M.; Yeung, K.S.; Wong, W.L.; Chung, B.H.; Tong, T.M.; Lo, I.F. Silver-Russell syndrome in Hong Kong. Hong Kong Med. J. 2016, 22, 526–533. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, S.Y.; Shin, C.H.; Lee, Y.A.; Shin, C.H.; Yang, S.W.; Cho, T.J.; Ko, J.M. Clinical Application of Sequential Epigenetic Analysis for Diagnosis of Silver-Russell Syndrome. Ann. Lab. Med. 2021, 41, 401–408. [Google Scholar] [CrossRef]
- Wakeling, E.L.; Amero, S.A.; Alders, M.; Bliek, J.; Forsythe, E.; Kumar, S.; Lim, D.H.; MacDonald, F.; Mackay, D.J.; Maher, E.R.; et al. Epigenotype-phenotype correlations in Silver-Russell syndrome. J. Med. Genet. 2010, 47, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amero, S.; Monk, D.; Frost, J.; Preece, M.; Stanier, P.; Moore, G.E. The genetic aetiology of Silver-Russell syndrome. J. Med. Genet. 2008, 45, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Netchine, I.; Rossignol, S.; Dufourg, M.N.; Azzi, S.; Rousseau, A.; Perin, L.; Houang, M.; Steunou, V.; Esteva, B.; Thibaud, N.; et al. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell-Silver syndrome: Clinical scoring system and epigenetic-phenotypic correlations. J. Clin. Endocrinol. Metab. 2007, 92, 3148–3154. [Google Scholar] [CrossRef]
- Kotzot, D. Maternal uniparental disomy 7 and Silver-Russell syndrome—Clinical update and comparison with other subgroups. Eur. J. Med. Genet. 2008, 51, 444–451. [Google Scholar] [CrossRef]
- Price, S.M.; Stanhope, R.; Garrett, C.; Preece, M.A.; Trembath, R.C. The spectrum of Silver-Russell syndrome: A clinical and molecular genetic study and new diagnostic criteria. J. Med. Genet. 1999, 36, 837–842. [Google Scholar]
- Bartholdi, D.; Krajewska-Walasek, M.; Ounap, K.; Gaspar, H.; Chrzanowska, K.H.; Ilyana, H.; Kayserili, H.; Lurie, I.W.; Schinzel, A.; Baumer, A. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): Results from a large cohort of patients with SRS and SRS-like phenotypes. J. Med. Genet. 2009, 46, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Bliek, J.; Terhal, P.; van den Bogaard, M.J.; Maas, S.; Hamel, B.; Salieb-Beugelaar, G.; Simon, M.; Letteboer, T.; van der Smagt, J.; Kroes, H.; et al. Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am. J. Hum. Genet. 2006, 78, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, T. Russell-Silver syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hannula, K.; Kere, J.; Pirinen, S.; Holmberg, C.; Lipsanen-Nyman, M. Do patients with maternal uniparental disomy for chromosome 7 have a distinct mild Silver-Russell phenotype? J. Med. Genet. 2001, 38, 273–278. [Google Scholar] [CrossRef][Green Version]
- Fuke, T.; Mizuno, S.; Nagai, T.; Hasegawa, T.; Horikawa, R.; Miyoshi, Y.; Muroya, K.; Kondoh, T.; Numakura, C.; Sato, S.; et al. Molecular and clinical studies in 138 Japanese patients with Silver-Russell syndrome. PLoS ONE 2013, 8, e60105. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chang, M.H. New growth charts for Taiwanese children and adolescents based on World Health Organization standards and health-related physical fitness. Pediatr. Neonatol. 2010, 51, 69–79. [Google Scholar] [CrossRef]
- Lin, S.Y.; Lee, C.N.; Hung, C.C.; Tsai, W.Y.; Lin, S.P.; Li, N.C.; Hsieh, W.S.; Tung, Y.C.; Niu, D.M.; Hsu, W.M.; et al. Epigenetic profiling of the H19 differentially methylated region and comprehensive whole genome array-based analysis in Silver-Russell syndrome. Am. J. Med. Genet. A 2010, 152, 2521–2528. [Google Scholar] [CrossRef]
- Priolo, M.; Sparago, A.; Mammì, C.; Cerrato, F.; Laganà, C.; Riccio, A. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur. J. Hum. Genet. 2008, 16, 565–571. [Google Scholar] [CrossRef]
- Wojdacz, T.K.; Dobrovic, A.; Algar, E.M. Rapid detection of methylation change at H19 in human imprinting disorders using methylation-sensitive high-resolution melting. Hum. Mutat. 2008, 29, 1255–1260. [Google Scholar] [CrossRef]
- Matsubara, K.; Murakami, N.; Nagai, T.; Ogata, T. Maternal age effect on the development of Prader-Willi syndrome resulting from upd(15)mat through meiosis 1 errors. J. Hum. Genet. 2011, 56, 566–571. [Google Scholar] [CrossRef]
- Kopca, T.; Tulay, P. Association of Assisted Reproductive Technology Treatments with Imprinting Disorders. Glob. Med. Genet. 2021, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Hiura, H.; Kitamura, A.; Miyauchi, N.; Kobayashi, N.; Takahashi, S.; Okae, H.; Kyono, K.; Kagami, M.; Ogata, T.; et al. Association of four imprinting disorders and ART. Clin. Epigenet. 2019, 11, 21. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Classification | SRS Score (Maximum = 15) | Epigenetic and Genetic Defects | ||||
|---|---|---|---|---|---|---|
| IC1 Hypomethylation (%) | mUPD7 (%) | Microdeletion | Unknown (%) | Molecular Diagnosis Rate | ||
| Clinical diagnosis with SRS (score≥ 8) (n = 100) | 10.2 ± 1.9 | 36 (36%) | 7 (7%) | 2 (2%) | 55 (55%) | 45% |
| Subjects with SRS score ≤ 7 (n = 106) | 5.3 ± 1.5 | 1 (1%) | 3 (3%) | 1 (1%) | 101 (95%) | 5% |
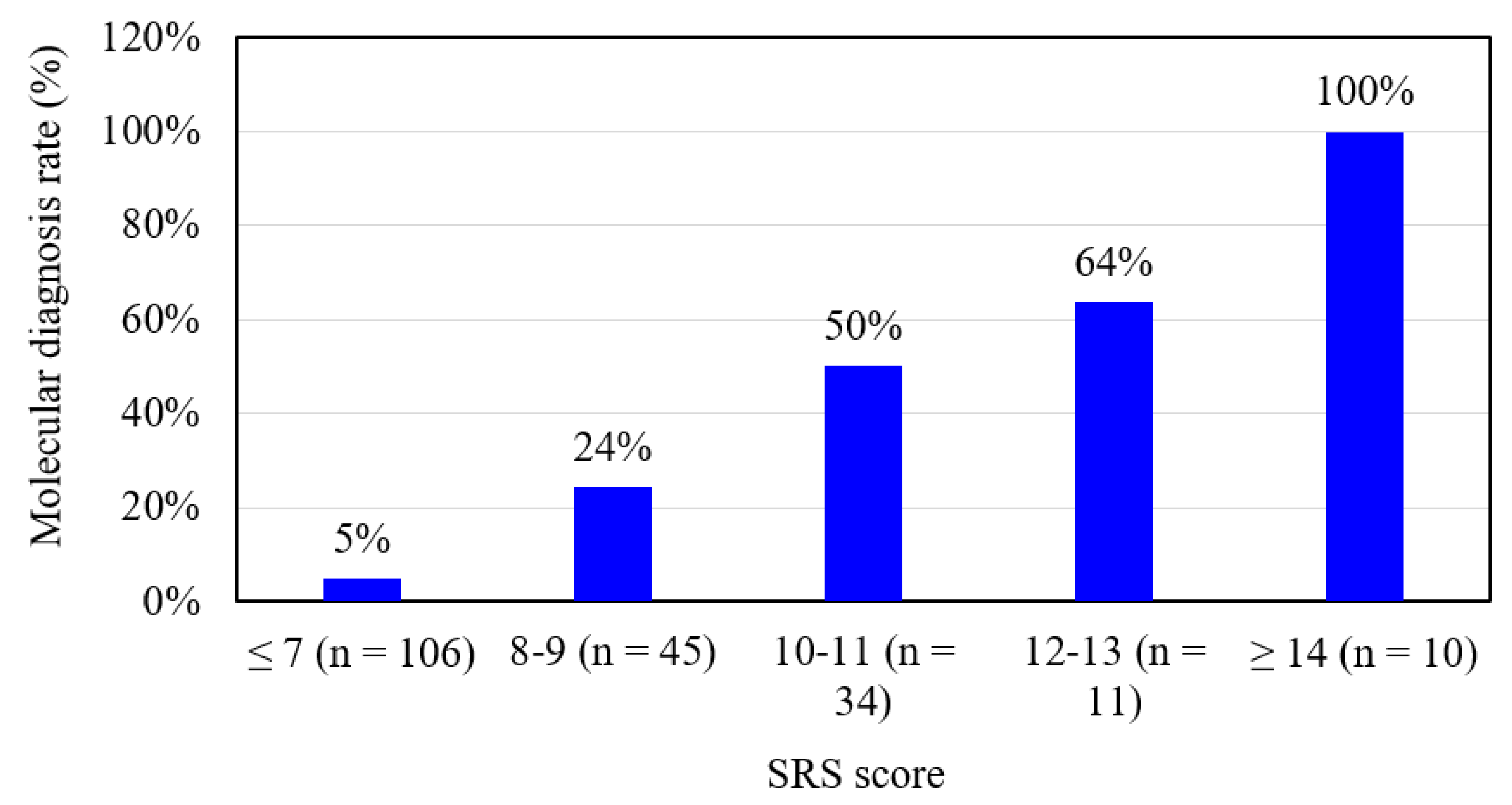
| SRS Score (Maximum = 15) | n | IC1 Hypomethylation | mUPD7 | Microdeletion | Unknown | Molecular Diagnosis | Molecular Diagnosis Rate (%) |
|---|---|---|---|---|---|---|---|
| ≤ 7 | 106 | 1 | 3 | 1 | 101 | 5 | 5% |
| 8 | 21 | 2 | 3 | 1 | 15 | 6 | 29% |
| 9 | 24 | 5 | 0 | 0 | 19 | 5 | 21% |
| 10 | 17 | 2 | 3 | 1 | 11 | 6 | 35% |
| 11 | 17 | 11 | 0 | 0 | 6 | 11 | 65% |
| 12 | 8 | 5 | 0 | 0 | 3 | 5 | 63% |
| 13 | 3 | 2 | 0 | 0 | 1 | 2 | 67% |
| 14 | 8 | 7 | 1 | 0 | 0 | 8 | 100% |
| 15 | 2 | 2 | 0 | 0 | 0 | 2 | 100% |
| All | 206 | 37 | 10 | 3 | 156 | 50 | 24% |
| Clinical Features | Clinical Diagnosis (n = 100) | p Value | Clinical Diagnosis (n = 100) | Netchine et al. [8] (n = 39) | |
|---|---|---|---|---|---|
| With Identified Molecular Defect (n = 43) | Without Identified Molecular Defect (n = 57) | ||||
| Male/Female | 25/18 | 26/31 | 0.219 | 51/49 | 19/20 |
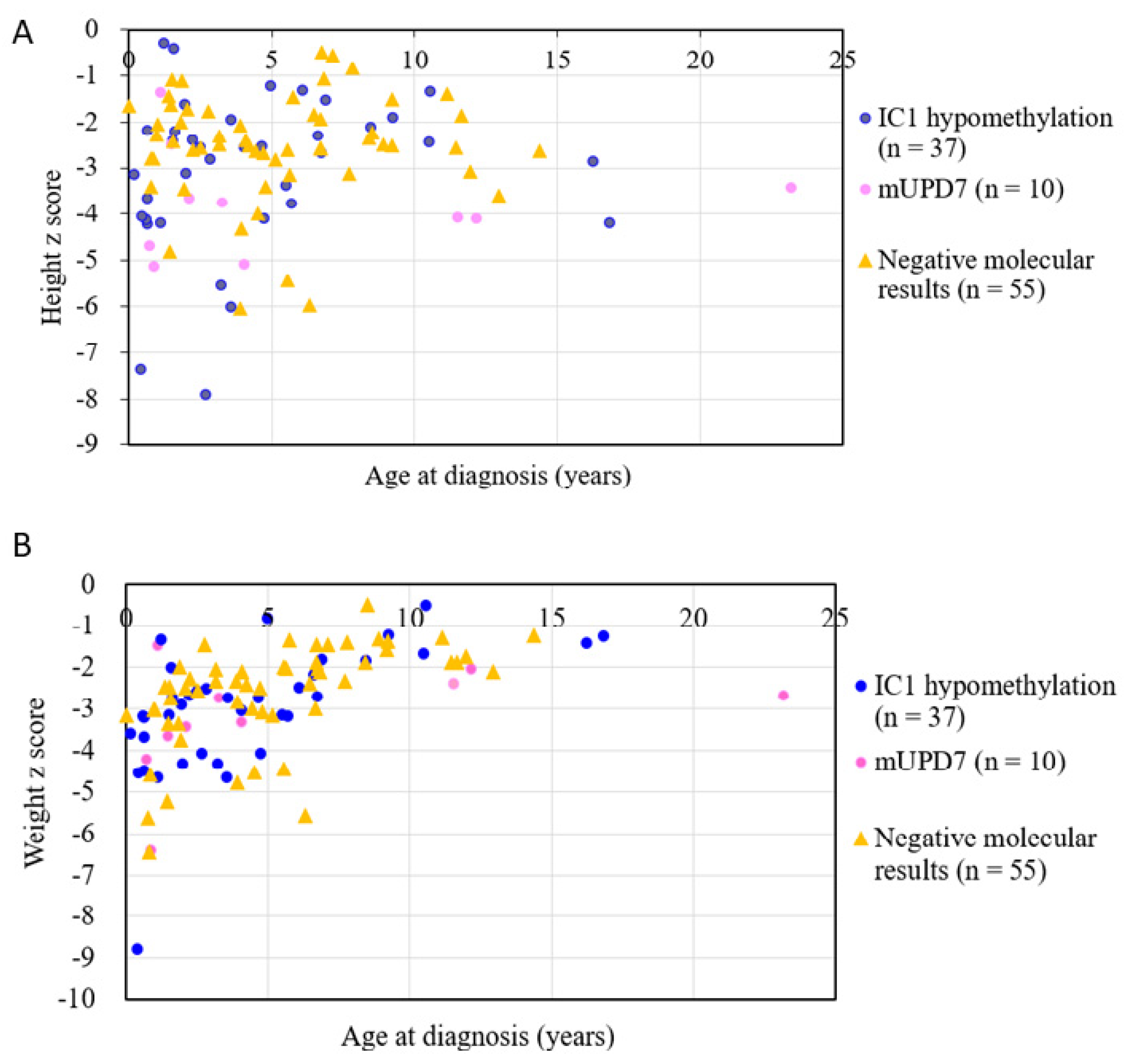
| Age at diagnosis (years) | 4.8 ± 5.0 | 5.2 ± 3.5 | 0.717 | 5.0 ± 4.2 | — |
| Height z score at diagnosis | −3.1 ± 1.6 | −2.6 ± 1.2 | 0.045 | −2.8 ± 1.4 | −3.6 ± 1.5 |
| Weight z score at diagnosis | −3.0 ± 1.4 | −2.7 ± 1.2 | 0.262 | −2.8 ± 1.3 | — |
| Gestational age (weeks) | 37.9 ± 1.7 (n = 41) | 37.3 ± 3.0 (n = 54) | 0.288 | 37.5 ± 2.5 (n = 95) | 37.5 ± 2.8 |
| Birth length z score | −4.1 ± 1.6 (n = 26) | −2.5 ± 1.4 (n = 38) | <0.001 | −2.7 ± 1.0 (n = 64) | −4.1 ± 1.5 |
| Birth weight z score | −3.0 ± 0.9 (n = 40) | −2.4 ± 1.0 (n = 54) | 0.009 | −3.1 ± 1.7 (n = 94) | −3.1 ± 1.2 |
| Birth OFC z score | −1.2 ± 2.6 (n = 22) | −1.8 ± 1.7 (n = 30) | 0.309 | −1.5 ± 2.1 (n = 52) | −1.5 ± 1.1 |
| Paternal age at childbirth (years) | 33.7 ± 4.8 (n = 29) | 34.4 ± 5.2 (n = 40) | 0.590 | 34.1 ± 5.0 (n = 69) | — |
| Maternal age at childbirth (years) | 31.3 ± 4.8 (n = 29) | 31.7 ± 5.1 (n = 40) | 0.755 | 31.6 ± 4.9 (n = 69) | — |
| Parameters at birth | |||||
| Weight ≤ 10th centile | 98% | 86% | 0.043 | 91% | — |
| Length ≤ 10th centile | 98% | 84% | 0.026 | 90% | — |
| Relative macrocephaly | 86% | 33% | <0.001 | 56% | 84.6% |
| Postnatal course | |||||
| No catch-up growth; height ≤ 3rd centile | 93% | 88% | 0.387 | 90% | 92.3% |
| Normal head circumference; OFC ≥ 3rd centile and ≤97th centile | 88% | 60% | 0.001 | 72% | — |
| Normal cognitive development | 86% | 68% | 0.041 | 76% | 76.9% |
| Asymmetry | |||||
| Face/body/limbs | 77% | 56% | 0.033 | 65% | 64.1% |
| Facial features | |||||
| Triangular shaped face | 84% | 88% | 0.573 | 86% | — |
| High/bossing forehead | 84% | 79% | 0.552 | 81% | 92.3% |
| Other: e.g., small chin, thin lips, downturned corners of the mouth, late closure of fontanelle | 67% | 65% | 0.794 | 66% | — |
| Other features | |||||
| Clinodactyly of the fifth finger | 63% | 63% | 0.970 | 63% | 64.1% |
| Genital abnormalities (e.g., cryptorchidism, hypospadias) | 16% | 14% | 0.759 | 15% | — |
| Other: e.g., brachymesophalangy, syndactyly toes, inguinal hernia, pigmentary changes | 33% | 40% | 0.429 | 37% | 17.9% |
| Total score (maximum = 15) | 11.3 ± 2.1 | 9.4 ± 1.2 | <0.001 | 10.2 ± 1.9 | — |
| Clinical Features | IC1 Hypomethylation (n = 37) | mUPD7 (n = 10) | p Value |
|---|---|---|---|
| Male/Female | 23/14 | 4/6 | 0.217 |
| Age at diagnosis (years) | 4.4 ± 4.1 | 6.1 ± 7.4 | 0.355 |
| Height z score at diagnosis | −3.0 ± 1.7 | −3.8 ± 1.2 | 0.199 |
| Weight z score at diagnosis | −3.0 ± 1.5 | −3.2 ± 1.4 | 0.639 |
| Gestational age (weeks) | 38.0 ± 1.7 (n = 35) | 36.6 ± 1.6 (n = 9) | 0.026 |
| Birth length z score | −4.1 ± 1.8 (n = 23) | −3.7 ± 0.9 (n = 5) | 0.578 |
| Birth weight z score | −3.0 ± 0.9 (n = 34) | −2.5 ± 0.9 (n = 9) | 0.150 |
| Birth OFC z score | −1.3 ± 1.3 (n = 20) | −1.6 ± 1.1 (n = 4) | 0.800 |
| Paternal age at childbirth (years) | 32.9 ± 4.3 (n = 25) | 37.6 ± 4.8 (n = 7) | 0.018 |
| Maternal age at childbirth (years) | 30.3 ± 4.1 (n = 25) | 35.3 ± 5.1 (n = 7) | 0.011 |
| Parameters at birth | |||
| Weight ≤ 10th centile | 97% | 100% | 0.609 |
| Length ≤ 10th centile | 100% | 90% | 0.053 |
| Relative macrocephaly | 89% | 50% | 0.004 |
| Postnatal course | |||
| No catch-up growth; height ≤ 3rd centile | 92% | 100% | 0.363 |
| Normal head circumference; OFC ≥ 3rd centile and ≤ 97th centile | 89% | 80% | 0.451 |
| Normal cognitive development | 86% | 80% | 0.618 |
| Asymmetry | |||
| Face/body/limbs | 76% | 50% | 0.120 |
| Facial features | |||
| Triangular shaped face | 89% | 50% | 0.004 |
| High/bossing forehead | 81% | 80% | 0.940 |
| Other: e.g., small chin, thin lips, downturned corners of the mouth, late closure of fontanelle | 68% | 60% | 0.662 |
| Other features | |||
| Clinodactyly of the fifth finger | 68% | 20% | 0.006 |
| Genital abnormalities (e.g., cryptorchidism, hypospadias) | 19% | 0% | 0.142 |
| Others: e.g., brachymesophalangy, syndactyly toes, inguinal hernia, pigmentary changes | 35% | 10% | 0.128 |
| Total score (maximum = 15) | 11.4 ± 2.2 | 8.7 ± 2.5 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-Y.; Lee, C.-L.; Fran, S.; Tu, R.-Y.; Chang, Y.-H.; Niu, D.-M.; Chang, C.-Y.; Chiu, P.-C.; Chou, Y.-Y.; Hsiao, H.-P.; et al. Epigenotype, Genotype, and Phenotype Analysis of Taiwanese Patients with Silver–Russell Syndrome. J. Pers. Med. 2021, 11, 1197. https://doi.org/10.3390/jpm11111197
Lin H-Y, Lee C-L, Fran S, Tu R-Y, Chang Y-H, Niu D-M, Chang C-Y, Chiu P-C, Chou Y-Y, Hsiao H-P, et al. Epigenotype, Genotype, and Phenotype Analysis of Taiwanese Patients with Silver–Russell Syndrome. Journal of Personalized Medicine. 2021; 11(11):1197. https://doi.org/10.3390/jpm11111197
Chicago/Turabian StyleLin, Hsiang-Yu, Chung-Lin Lee, Sisca Fran, Ru-Yi Tu, Ya-Hui Chang, Dau-Ming Niu, Chia-Ying Chang, Pao-Chin Chiu, Yen-Yin Chou, Hui-Pin Hsiao, and et al. 2021. "Epigenotype, Genotype, and Phenotype Analysis of Taiwanese Patients with Silver–Russell Syndrome" Journal of Personalized Medicine 11, no. 11: 1197. https://doi.org/10.3390/jpm11111197
APA StyleLin, H.-Y., Lee, C.-L., Fran, S., Tu, R.-Y., Chang, Y.-H., Niu, D.-M., Chang, C.-Y., Chiu, P.-C., Chou, Y.-Y., Hsiao, H.-P., Tsai, M.-C., Chao, M.-C., Tsai, L.-P., Yang, C.-F., Su, P.-H., Pan, Y.-W., Lee, C.-H., Chu, T.-H., Chuang, C.-K., & Lin, S.-P. (2021). Epigenotype, Genotype, and Phenotype Analysis of Taiwanese Patients with Silver–Russell Syndrome. Journal of Personalized Medicine, 11(11), 1197. https://doi.org/10.3390/jpm11111197