
Xeroderma Pigmentosum: General Aspects and Management

 ,
,  , ,
, , 
Abstract
1. Introduction
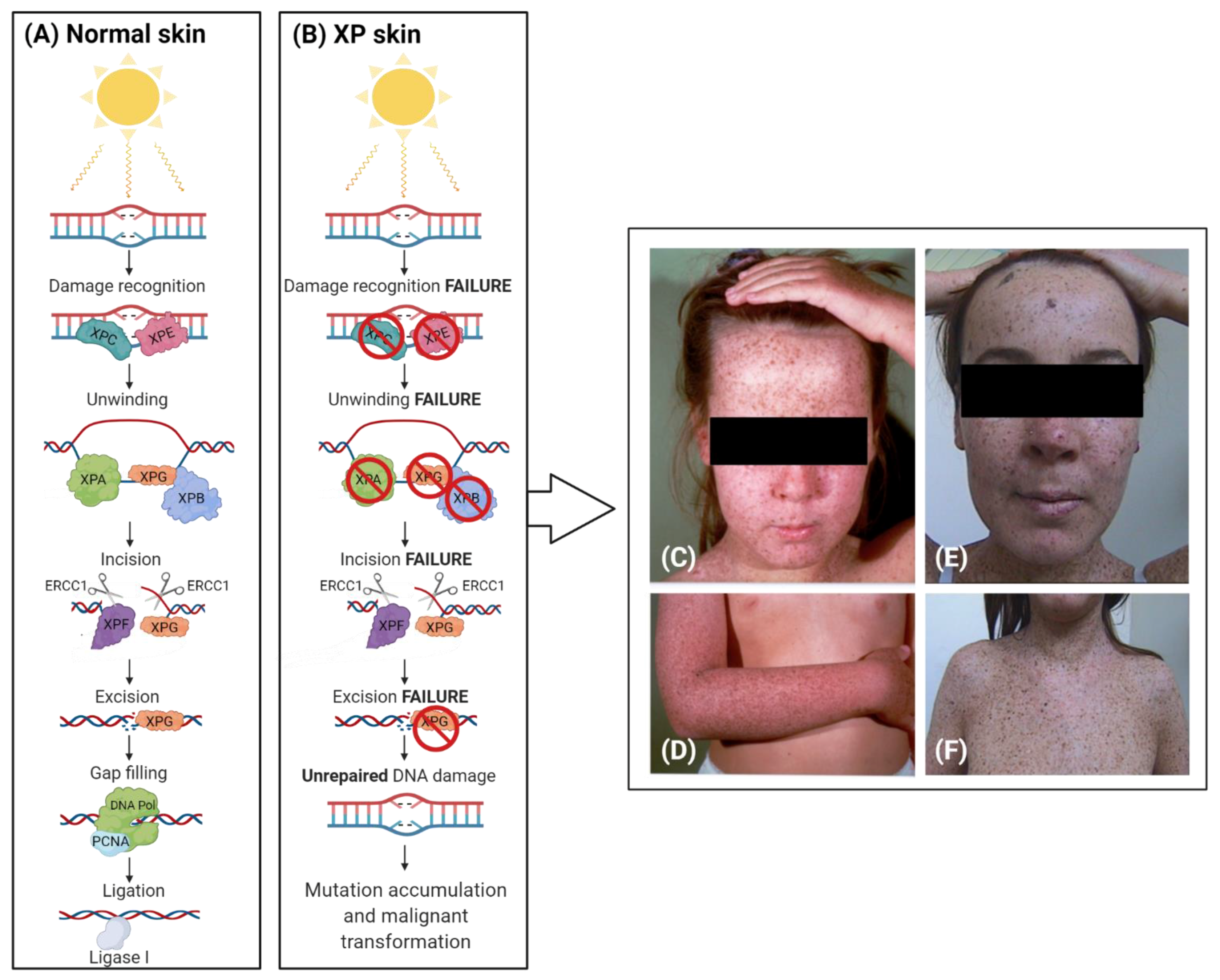
Clinical Features of Xeroderma Pigmentosum

{kind=link}
{kind=link}
{kind=link}
| Group | Gravity Score [11] | Photosensitivity (Sunburn) | Xerosis | Pigmentary Abnormalities | Increased Skin Cancer Risk | Neurological Disorders | Eye-Disorders |
|---|---|---|---|---|---|---|---|
| XP-A | M/S | +/−[13] | +[14] | +[13] | +[11] | +[15] | +[14] |
| XP-B | M | +[16] | +[16] | +[16] | +[11,16,17] | +[6,18,19] | +[19] |
| XP-C | M/S | +[20] | +[20,21] | +[20,21] | +[11,20,22,23] | −[6,9,20,24] | +[20] |
| XP-D | M | +[25] | +[26] | +[25] | +[11] | +[6,18,27] | +[6] |
| XP-E | M | +[2] | +[2] | +[2] | +/−[11,28] | −[6] | +[6] |
| XP-F | V | +[29] | +[29] | +[29] | +[11] | −[6,30] | +[6] |
| XP-G | M/S | +[26,31] | +[26] | +[2] | +[11,32] | +[6,18,31] | +[6] |
| XP-V | V | +/−[10,18] | +[33] | +/−[18] | −[11] | −[6] | +[6,34] |
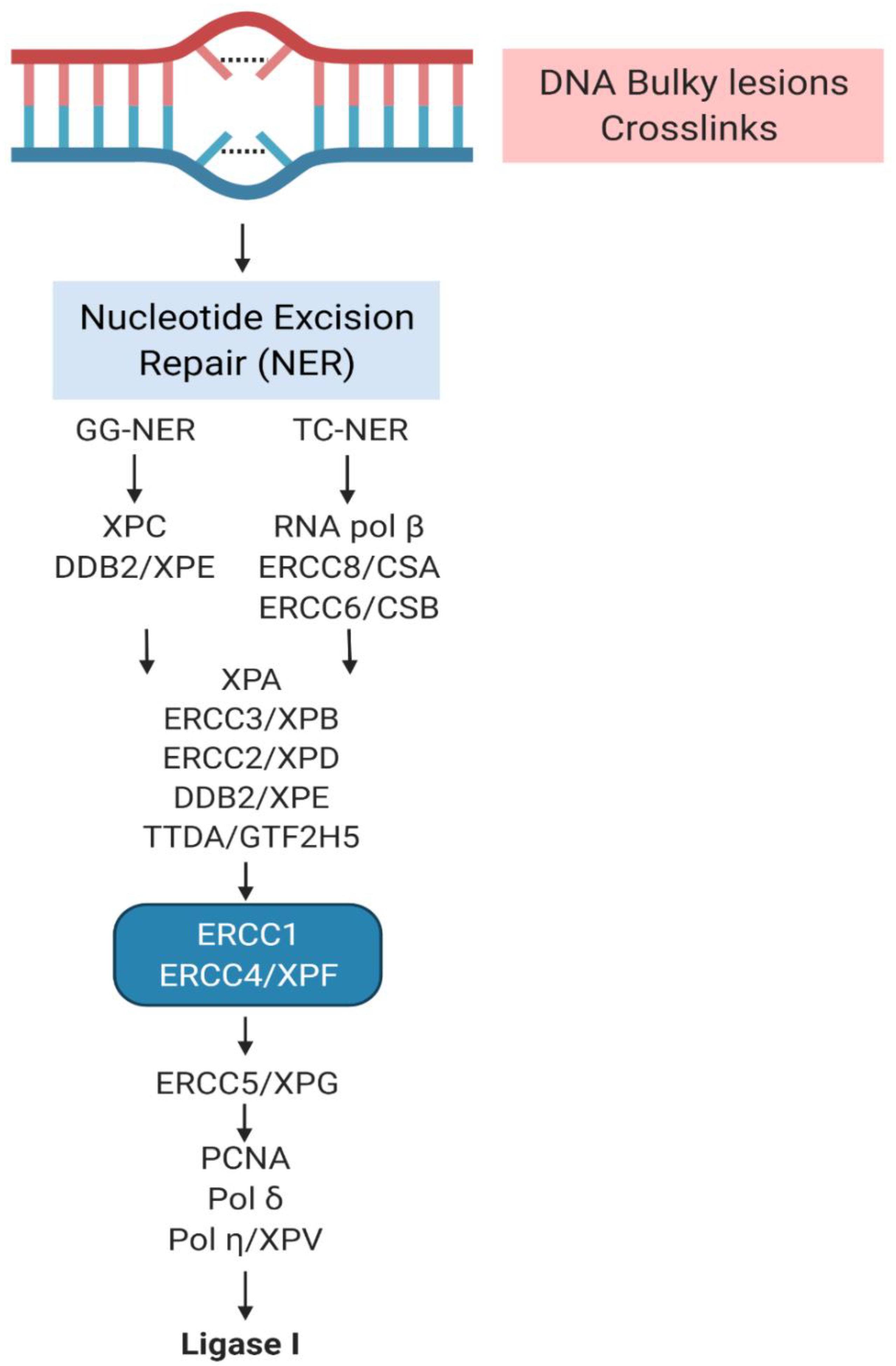
2. The Complex Involvement of NER
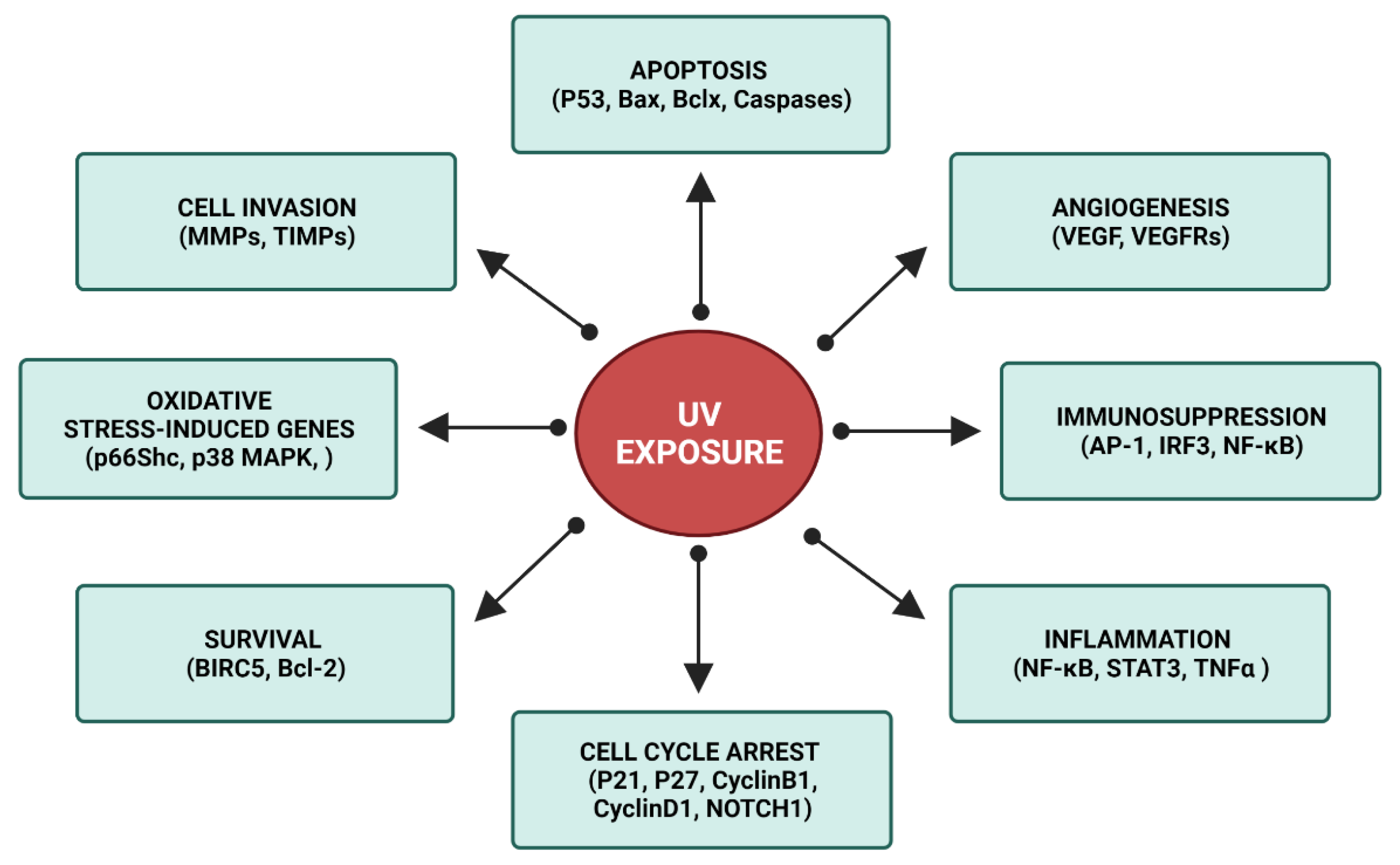
3. XP Molecular Insights
4. First-Generation Intervention Strategies
4.1. Physical/Chemical Photoprotectors
4.2. Dermabrasion and Chemical Peeling
4.3. Administration of Retinoids
4.4. Phototherapy and Laser Resurfacing
4.5. Chemotherapeutic Drugs
5. Next Generation Approaches: Target Therapies
5.1. Gene Therapy and Autologous Transplantation
5.2. Liposomal Formulations as Nanocarriers for DNA Repair Enzymes
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bradford, P.T.; Goldstein, A.M.; Tamura, D.; Khan, S.; Ueda, T.; Boyle, J.; Oh, K.-S.; Imoto, K.; Inui, H.; Moriwaki, S.I.; et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011, 48, 168–176. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a Light on Xeroderma Pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef]
- Brem, R.; Macpherson, P.; Guven, M.; Karran, P. Oxidative stress induced by UVA photoactivation of the tryptophan UVB photoproduct 6-formylindolo[3,2-b]carbazole (FICZ ) inhibits nucleotide excision repair in human cells. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.orphadata.org/cgi-bin/index.php (accessed on 10 September 2021).
- De Boer, J.; Hoeijmakers, J.H.J. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Hengge, U.R.; Emmert, S. Clinical features of xeroderma pigmentosum. Adv. Exp. Med. Biol. 2008, 637, 10–18. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Lee, M.; Andrews, A.D.; Lambert, W.C. The Role of Sunlight and DNA Repair in Melanoma and Nonmelanoma Skin Cancer-The Xeroderma Pigmentosum Paradigm. Arch. Dermatol. 1994, 130, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Caso, E.R.; Marcos, A.A.; Morales, M.; Belfort, R.N. Simultaneous squamous cell carcinoma and malignant melanoma of the conjunctiva in a teenager with xeroderma pigmentosum: Case report. Indian J. Ophthalmol. 2019, 67, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Sethi, M.; Lehmann, A.R.; Fassihi, H. Xeroderma Pigmentosum: A Multidisciplinary Approach. EMJ Dermatol. 2013, 1, 54–63. [Google Scholar]
- Black, J.O. Xeroderma Pigmentosum. Head Neck Pathol. 2016, 10, 139–144. [Google Scholar] [CrossRef]
- Zghal, M.; Fazaa, B.; Abdelhak, S.; Mokni, M. Xeroderma pigmentosum. Ann. Dermatol. Venereol. 2018, 145, 706–722. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, D.; Hoeijmakers, J.H.J. The genetic basis of xeroderma pigmentosum. Ann. Genet. 1991, 34, 143–150. [Google Scholar] [PubMed]
- Salob, S.P.; Webb, D.K.H.; Atherton, D.J. A child with xeroderma pigmentosum and bone marrow failure. Br. J. Dermatol. 1992, 126, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Santiago, K.M.; De Nóbrega, A.F.; Rocha, R.M.; Rogatto, S.R.; Achatz, M.I. Xeroderma pigmentosum: Low prevalence of germline XPA mutations in a brazilian XP population. Int. J. Mol. Sci. 2015, 16, 8988–8996. [Google Scholar] [CrossRef]
- Kanda, T.; Oda, M.; Yonezawa, M.; Tamagawa, K.; Isa, F.; Hanakago, R.; Tsukagoshi, H. Peripheral neuropathy in xeroderma pigmentosum. Brain 1990, 113, 1025–1044. [Google Scholar] [CrossRef]
- Oh, K.S.; Khan, S.; Jaspers, N.G.J.; Raams, A.; Ueda, T.; Lehmann, A.; Friedmann, P.S.; Emmert, S.; Gratchev, A.; Lachlan, K.; et al. Phenotypic Heterogeneity in the XPB DNA Helicase Gene (ERCC3): Xeroderma Pigmentosum Without and with Cockayne Syndrome. Hum. Mutat. 2006, 27, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.H.; Kraemer, K.H.; Lutzner, M.A.; Festoff, B.W.; Coon, H.G. Xeroderma Pigmentosum An Inherited Disease with Sun Sensitivity, Multiple Cutaneous Neoplasms, and Abnormal DNA Repair. Ann. Intern. Med. 1974, 80, 221–248. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, S.; Kanda, F.; Hayashi, M.; Yamashita, D.; Sakai, Y.; Nishigori, C. Xeroderma pigmentosum clinical practice guidelines. J. Dermatol. 2017, 44, 1087–1096. [Google Scholar] [CrossRef]
- Scott, R.J.; Itin, P.; Kleijer, W.J.; Kolb, K.; Arlett, C.; Muller, H. Xeroderma pigmentosum-Cockayne syndrome complex in two patients: Absence of slun tumors despite severe deficiency of DNA excision repair. J. Am. Acad. Dermatol. 1993, 29, 883–889. [Google Scholar] [CrossRef]
- Hadj-Rabia, S.; Oriot, D.; Soufir, N.; Dufresne, H.; Bourrat, E.; Mallet, S.; Poulhalon, N.; Ezzedine, K.; Grandchamp, B.; Taïeb, A.; et al. Unexpected extradermatological findings in 31 patients with xeroderma pigmentosum type C. Br. J. Dermatol. 2013, 168, 1109–1113. [Google Scholar] [CrossRef]
- Gozukara, E.M.; Khan, S.; Metin, A.; Emmert, S.; Busch, D.B.; Shahlavi, T.; Coleman, D.M.; Miller, M.; Chinsomboon, N.; Stefanini, M.; et al. A stop codon in xeroderma pigmentosum group C families in Turkey and Italy: Molecular genetic evidence for a common ancestor. J. Investig. Dermatol. 2001, 117, 197–204. [Google Scholar] [CrossRef][Green Version]
- Lynch, H.T.; Fusaro, R.M.; Johnson, J.A. Xeroderma pigmentosum: Complementation group C and malignant melanoma. Arch. Derm. 1984, 120, 175–179. [Google Scholar] [CrossRef]
- Khan, S.G. Two essential splice lariat branchpoint sequences in one intron in a xeroderma pigmentosum DNA repair gene: Mutations results in reduced XPC mRNA levels that correlate with cancre risk. Hum. Mol. Genet. 2004, 13, 343–352. [Google Scholar] [CrossRef]
- Chavanne, F.; Broughton, B.C.; Pietra, D.; Nardo, T.; Browitt, A.; Lehmann, A.R.; Stefanini, M. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein, and transcript levels. Cancer Res. 2000, 60, 1974–1982. [Google Scholar] [PubMed]
- The UniProt Consortium. Disease-Xeroderma Pigmentosum Complementation Group D. Available online: https://www.uniprot.org/diseases/1158 (accessed on 10 September 2021).
- Moriwaki, S.; Takigawa, M.; Igarashi, N.; Nagai, Y.; Amano, H.; Ishikawa, O.; Khan, S.; Kraemer, K. Xeroderma pigmentosum complementation group G patient with a novel homozygous missense mutation and no neurological abnormalities. Exp. Dermatol. 2012, 21, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.T.; Squires, S. The XPD complementation group: Insights into xeroderma pigmentosum, Cockayne’s syndrome and trichothiodystrophy. Mutat. Res. 1992, 273, 97–118. [Google Scholar] [CrossRef]
- Chu, G.; Chang, E. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 1988, 242, 564–567. [Google Scholar] [CrossRef]
- Norris, P.G.; Hawk, J.L.M.; Avery, J.A.; Giannelli, F. Xeroderma pigmentosum complementation group F in a non-Japanese patient. J. Am. Acad. Dermatol. 1988, 18, 1185–1188. [Google Scholar] [CrossRef]
- Yamamura, K.; Ichihashi, M.; Hiramoto, T.; Ogoshi, M.; Nishioka, K.; Fujiwara, Y. Clinical and photobiological characteristics of xeroderma pigmentosum complementation group F: A review of cases from Japan. Br. J. Dermatol. 1989, 121, 471–480. [Google Scholar] [CrossRef]
- Chessbrough, M. Xeroderma pigmentosum––A unique variant with neurological involvement. Br. J. Dermatol. 1978, 99, 61. [Google Scholar] [CrossRef]
- Zafeiriou, D.; Thorel, F.; Andreou, A.; Kleijer, W.J.; Raams, A.; Garritsen, V.H.; Gombakis, N.; Jaspers, N.G.J.; Clarkson, S.G. Xeroderma pigmentosum group G with severe neurological involvement and features of Cockayne syndrome in infancy. Pediatr. Res. 2001, 49, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Lambert, W.C.; Lambert, M.W. Development of effective skin cancer treatment and prevention in xeroderma pigmentosum. Photochem. Photobiol. 2015, 91, 475–483. [Google Scholar] [CrossRef]
- Schelini, M.C.; Chaves, L.F.O.B.; Toledo, M.C.; Rodrigues, F.W.; de Oliveira, T.; Isaac, D.L.C.; Avila, M. Xeroderma Pigmentosum: Ocular Findings in an Isolated Brazilian Group with an Identified Genetic Cluster. J. Ophthalmol. 2019, 2019, 4818162. [Google Scholar] [CrossRef]
- Martires, K.J.; Fu, P.; Polster, A.M.; Cooper, K.D.; Baron, E.D. Factors That Affect Skin Aging. Arch. Dermatol. 2009, 145, 1375–1379. [Google Scholar] [CrossRef]
- Parrado, C.; Mercado-Saenz, S.; Perez-Davo, A.; Gilaberte, Y.; Gonzalez, S.; Juarranz, A. Environmental Stressors on Skin Aging. Mechanistic Insights. Front. Pharmacol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Heck, D.E.; Gerecke, D.R.; Vetrano, A.M.; Laskin, J.D. Solar ultraviolet radiation as a trigger of cell signal transduction. Toxicol. Appl. Pharmacol. 2004, 195, 288–297. [Google Scholar] [CrossRef]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef]
- Lee, C.; Wu, S.; Hong, C.; Yu, H.; Wei, Y. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. Int. J. Mol. Sci. 2013, 14, 6414–6435. [Google Scholar] [CrossRef]
- Zheng, J.; Mo, H.-Y.; Wang, Z.-Z. Clinicopathological characteristics of xeroderma pigmentosum associated with keratoacanthoma: A case report and literature review. Int. J. Clin. Exp. Med. 2014, 7, 3410–3414. [Google Scholar]
- de Feraudy, S.; Boubakour-Azzouz, I.; Fraitag, S.; Berneburg, M.; Chan, L.; Chew, K.; Clericuzio, C.L.; Cunningham, B.; Tope, W.D.; Cleaver, J.E. Diagnosing Xeroderma pigmentosum group C by immunohistochemistry. Am. J. Dermatopathol. 2010, 32, 109–117. [Google Scholar] [CrossRef]
- Stokes, M.P.; Comb, M.J. A wide-ranging cellular response to UV damage of DNA. Cell Cycle 2008, 7, 2097–2099. [Google Scholar] [CrossRef]
- Gentile, M.; Latonen, L.; Laiho, M. Cell cycle arrest and apoptosis provoked by UV radiation-induced DNA damage are transcriptionally highly divergent responses. Nucleic Acids Res. 2003, 31, 4779–4790. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Selby, C.P.; Adar, S.; Adebali, O.; Sancar, A. Molecular mechanisms and genomic maps of DNA excision repair in Escherichia coli and humans. J. Biol. Chem. 2017, 292, 15588–15597. [Google Scholar] [CrossRef]
- Gillet, L.C.J.; Schärer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Roger, A.S.; Wolfram, S.; Graham, C.W.; Tom, E.; Richard, D.W. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006; Available online: https://books.google.it/books/about/DNA_Repair_and_Mutagenesis.html?id=xVmZKUie2lkC&redir_esc=y (accessed on 10 September 2021).
- Fassihi, H. Spotlight on ’xeroderma pigmentosum’. Photochem. Photobiol. Sci. 2013, 12, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, O.; Kanaar, R. Dealing with DNA damage: Relationships between checkpoint and repair pathways. Mutat. Res. 2010, 704, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Geacintov, N.E.; Broyde, S.; Yeo, J.E.; Schärer, O.D. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair (Amst) 2018, 71, 33–42. [Google Scholar] [CrossRef]
- Kusakabe, M.; Onishi, Y.; Tada, H.; Kurihara, F.; Kusao, K.; Furukawa, M.; Iwai, S.; Yokoi, M.; Sakai, W.; Sugasawa, K. Mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair. Genes Environ. 2019, 41, 1–6. [Google Scholar] [CrossRef]
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Petruseva, I.O.; Lavrik, O.I. Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucleic Acids Res. 2010, 38, 8083–8094. [Google Scholar] [CrossRef]
- Sugitani, N.; Sivley, R.M.; Perry, K.E.; Capra, J.A.; Chazin, W.J. XPA: A key scaffold for human NER. DNA Repair 2016, 176, 139–148. [Google Scholar] [CrossRef]
- Beckwitt, E.C.; Jang, S.; Detweiler, I.C.; Kuper, J.; Sauer, F.; Simon, N.; Bretzler, J.; Watkins, S.C.; Carell, T.; Kisker, C.; et al. Single molecule analysis reveals monomeric XPA bends DNA and undergoes episodic linear diffusion during damage search. Nat. Commun. 2020, 11, 1356. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Weng, Q.Y.; Fisher, D.E. UV signaling pathways within the skin. J. Investig. Dermatol. 2014, 134, 2080–2085. [Google Scholar] [CrossRef]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Schärer, O.D.; Wagner, G.; Ellenberger, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef]
- Fagbemi, A.F.; Orelli, B.; Schärer, O.D. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair (Amst) 2011, 10, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Reardon, J.T.; Lindsey-Boltz, L.A.; Sancar, A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 2012, 287, 22889–22899. [Google Scholar] [CrossRef] [PubMed]
- Krasikova, Y.; Rechkunova, N.; Lavrik, O. Nucleotide excision repair: From molecular defects to neurological abnormalities. Int. J. Mol. Sci. 2021, 22, 6220. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–19. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, D.H.; Shin, M.H.; Shin, H.S.; Kim, M.K.; Chung, J.H. UV-induced DNA methyltransferase 1 promotes hypermethylation of tissue inhibitor of metalloproteinase 2 in the human skin. J. Dermatol. Sci. 2018, 91, 19–27. [Google Scholar] [CrossRef]
- Zhu, J.W.; Wu, X.J.; Lu, Z.F.; Luo, D.; Cai, S.Q.; Zheng, M. Role of VEGF Receptors in Normal and Psoriatic Human Keratinocytes: Evidence from Irradiation with Different UV Sources. PLoS ONE 2013, 8, e55463. [Google Scholar] [CrossRef] [PubMed]
- Trompezinski, S.; Pernet, I.; Schmitt, D.; Viac, J. UV radiation and prostaglandin E2 up-regulate vascular endothelial growth factor (VEGF) in cultured human fibroblasts. Inflamm. Res. 2001, 50, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zheng, Y.W.; Liu, Q.; Liu, L.P.; Luo, F.L.; Zhou, H.C.; Isoda, H.; Ohkohchi, N.; Li, Y.M. Reactive Oxygen Species in Skin Repair, Regeneration, Aging, and Inflammation. In Reactive Oxygen Species (ROS) in Living Cells; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Singh, A.; Willems, E.; Singh, A.; Bin Hafeez, B.; Ong, I.M.; Mehta, S.L.; Verma, A.K. Ultraviolet radiation-induced tumor necrosis factor alpha, which is linked to the development of cutaneous SCC, modulates differential epidermal microRNAs expression. Oncotarget 2016, 7, 17945–17956. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Ratnakumar, K.; Hung, K.-F.; Rokunohe, D.; Kawasumi, M. Deciphering UV-induced DNA responses to prevent and treat skin cancer. Photochem. Photobiol. 2020, 96, 478–499. [Google Scholar] [CrossRef]
- Podmirseg, S.R.; Jäkel, H.; Ranches, G.D.; Kullmann, M.K.; Sohm, B.; Villunger, A.; Lindner, H.; Hengst, L. Caspases uncouple p27Kip1 from cell cycle regulated degradation and abolish its ability to stimulate cell migration and invasion. Oncogene 2016, 35, 4580–4590. [Google Scholar] [CrossRef][Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Saltarella, I.; Vinella, A.; Muzio, L.L.; Pannone, G.; Fumarulo, R.; Vacca, A.; Mariggiò, M.A. Survivin overexpression in head and neck squamous cell carcinomas as a new therapeutic target (Review). Oncol. Rep. 2019, 41, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Hasan Aziz, M.; Ghotra, A.S.; Shukla, Y.; Ahmad, N. Ultraviolet-B Radiation Causes an Upregulation of Survivin in Human Keratinocytes and Mouse Skin. Photochem. Photobiol. 2004, 80, 602. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ananthaswamy, H.N. Toxic effects of ultraviolet radiation on the skin. Toxicol. Appl. Pharmacol. 2004, 195, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T.; Jacobson, M.K.; Jacobson, E.L. Endogenous UVA-photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection. Photochem. Photobiol. Sci. 2006, 5, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res. 2005, 571, 19–31. [Google Scholar] [CrossRef] [PubMed]
- You, Y.H.; Lee, D.H.; Yoon, J.H.; Nakajima, S.; Yasui, A.; Pfeifer, G.P. Cyclobutane Pyrimidine Dimers Are Responsible for the Vast Majority of Mutations Induced by UVB Irradiation in Mammalian Cells. J. Biol. Chem. 2001, 276, 44688–44694. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Nairn, R.S. The biology of the (6-4) photoproduct. Photochem. Photobiol. 1989, 49, 805–819. [Google Scholar] [CrossRef]
- Yokoyama, H.; Mizutani, R. Structural biology of DNA (6-4) photoproducts formed by ultraviolet radiation and interactions with their binding proteins. Int. J. Mol. Sci. 2014, 15, 20321–20338. [Google Scholar] [CrossRef] [PubMed]
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.; Halaban, R.; Douki, T.; Brash, D.E. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Vink, A.A.; Roza, L. Biological consequences of cyclobutane pyrimidine dimers. J. Photochem. Photobiol. B Biol. 2001, 65, 101–104. [Google Scholar] [CrossRef]
- Pedeux, R.; Al-Irani, N.; Marteau, C.; Pellicier, F.; Branche, R.; Ozturk, M.; Franchi, J.; Doré, J.F. Thymidine dinucleotides induce S phase cell cycle arrest in addition to increased melanogenesis in human melanocytes. J. Investig. Dermatol. 1998, 111, 472–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abeyama, K.; Eng, W.; Jester, J.V.; Vink, A.A.; Edelbaum, D.; Cockerell, C.J.; Bergstresser, P.R.; Takashima, A. A role for NF-κB-dependent gene transactivation in sunburn. J. Clin. Investig. 2000, 105, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.H.; Chien, A.L. Photoaging: A Review of Current Literature. Curr. Dermatol. Rep. 2020, 9, 22–29. [Google Scholar] [CrossRef]
- Vierkötter, A.; Krutmann, J. Environmental influences on skin aging and ethnic-specific manifestations. Dermatoendocrinology 2012, 4, 227–231. [Google Scholar] [CrossRef] [PubMed]
- De Zio, D.; Cianfanelli, V.; Cecconi, F. New insights into the link between DNA damage and apoptosis. Antioxidants Redox Signal. 2013, 19, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, D.B.; Rosenthal, A.; Moy, R. Six critical questions for DNA repair enzymes in skincare products: A review in dialog. Clin. Cosmet. Investig. Dermatol. 2019, 12, 617–624. [Google Scholar] [CrossRef] [PubMed]
- López-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Pérez-Plasencia, C.; Álvarez-Sánchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142–172. [Google Scholar] [CrossRef] [PubMed]
- Perdiz, D.; Gróf, P.; Mezzina, M.; Nikaido, O.; Moustacchi, E.; Sage, E. Distribution and Repair of Bipyrimidine Photoproducts in Solar UV-irradiated Mammalian Cells. J. Biol. Chem. 2000, 275, 26732–26742. [Google Scholar] [CrossRef]
- Marionnet, C.; Armier, J.; Sarasin, A.; Stary, A. Cyclobutane pyrimidine dimers are the main mutagenic DNA photoproducts in DNA repair-deficient trichothiodystrophy cells. Cancer Res. 1998, 58, 102–108. [Google Scholar]
- De Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Adv. Exp. Med. Biol. 2017, 996, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Polefka, T.G.; Meyer, T.A.; Agin, P.P.; Bianchini, R.J. Effects of solar radiation on the skin. J. Cosmet. Dermatol. 2012, 11, 134–143. [Google Scholar] [CrossRef]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–925. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Alora-Palli, M.B.; Tamura, M.; Mullins, L.A.; Soh, C.; Binder, R.L.; Houston, N.A.; Conley, E.D.; Tung, J.Y.; Annunziata, N.E.; et al. Age-induced and photoinduced changes in gene expression profiles in facial skin of Caucasian females across 6 decades of age. J. Am. Acad. Dermatol. 2018, 78, 29–39.e7. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, N.K.; Varghese, M. Therapeutic Response of a Brother and Sister with Xeroderma Pigmentosum to Imiquimod 5% Cream. Dermatol. Surg. 2002, 28, 518–523. [Google Scholar] [CrossRef]
- Kugler, M. Xeroderma Pigmentosum Disease Symptoms and Treatment. 2019. Available online: https://www.verywellhealth.com/xeroderma-pigmentosum-2861056 (accessed on 10 September 2021).
- Latha, M.S.; Martis, J.; Shobha, V.; Shinde, R.S.; Bangera, S.; Krishnankutty, B.; Bellary, S.; Varughese, S.; Rao, P.; Naveen Kumar, B.R. Sunscreening agents: A review. J. Clin. Aesthet. Dermatol. 2013, 6, 16–26. [Google Scholar]
- Gabros, S.; Nessel, T.; Zito, P. Sunscreens and Photoprotection; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537164/ (accessed on 10 September 2021).
- Heurung, A.R.; Raju, S.I.; Warshaw, E.M. Adverse reactions to sunscreen agents: Epidemiology, responsible irritants and allergens, clinical characteristics, and management. Dermatitis 2014, 25, 289–326. [Google Scholar] [CrossRef]
- Milota, M.; Jones, D.L.; Cleaver, J.; Jamall, I.S. Xeroderma pigmentosum family support group: Helping families and promoting clinical initiatives. DNA Repair (Amst) 2011, 10, 792–797. [Google Scholar] [CrossRef]
- Tamura, D.; Digiovanna, J.J.; Khan, S.G.; Kraemer, K.H. Living with xeroderma pigmentosum: Comprehensive photoprotection for highly photosensitive patients. Photodermatol. Photoimmunol. Photomed. 2014, 30, 146–152. [Google Scholar] [CrossRef]
- Krause, M.; Klit, A.; Blomberg Jensen, M.; Søeborg, T.; Frederiksen, H.; Schlumpf, M.; Lichtensteiger, W.; Skakkebaek, N.E.; Drzewiecki, K.T. Sunscreens: Are they beneficial for health? An overview of endocrine disrupting properties of UV-filters. Int. J. Androl. 2012, 35, 424–436. [Google Scholar] [CrossRef]
- Jansen, R.; Osterwalder, U.; Wang, S.Q.; Burnett, M.; Lim, H.W. Photoprotection: Part II. Sunscreen: Development, efficacy, and controversies. J. Am. Acad. Dermatol. 2013, 69, 867.e1–867.e14. [Google Scholar] [CrossRef]
- Brescoll Mancuso, J.; Maruthi, R.; Wang, S.Q.; Lim, H.W. Sunscreens: An update. Am. J. Clin. Dermatol. 2017, 18, 643–650. [Google Scholar] [CrossRef]
- Smijs, T.; Pavel, S. Titanium dioxide and zinc oxide nanoparticles in sunscreens: Focus on their safety and effectiveness. Nanotechnol. Sci. Appl. 2011, 4, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Skotarczak, K.; Osmola-Mańkowska, A.; Lodyga, M.; Polańska, A.; Mazur, M.; Adamski, Z. Photoprotection: Facts and controversies. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 98–112. [Google Scholar]
- Wright, M.W.; Wright, S.T.; Wagner, R.F. Mechanisms of sunscreen failure. J. Am. Acad. Dermatol. 2001, 44, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Colantonio, S.; Dawson, A.; Lin, X.; Beecker, J. Sunscreen Application, Safety, and Sun Protection: The Evidence. J. Cutan. Med. Surg. 2019, 23, 357–369. [Google Scholar] [CrossRef]
- Directorate-European Commission Health & Consumer. Opinion on Benzophenone-3. COLIPA N° S38. 2016. Available online: https://ec.europa.eu/health/ph_risk/committees/04_sccp/docs/sccp_o_159.pdf (accessed on 10 September 2021).
- DiNardo, J.C.; Downs, C.A. Dermatological and environmental toxicological impact of the sunscreen ingredient oxybenzone/benzophenone-3. J. Cosmet. Dermatol. 2018, 17, 15–19. [Google Scholar] [CrossRef]
- Lin, P.; English III, J.C. Topical Treatment of Xeroderma Pigmentosum. Pharm. Ther. 2004, 29, 512–516. [Google Scholar]
- Gold, M.H. Dermabrasion in dermatology. Am. J. Clin. Dermatol. 2003, 4, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, K.; Veliath, A.J.; Mishra, S.; Panda, K.N. Xeroderma Pigmentosum: Resurfacing versus dermabrasion. Br. J. Plast. Surg. 1992, 45, 311–314. [Google Scholar] [CrossRef]
- Ocampo-Candiani, J.; Silva-Siwady, G.; Fernandez-Gutierrez, L.; Field, L.M. Dermabrasion in xeroderma pigmentosum. Dermatol. Surg. 1996, 22, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Alzacko, S.M.; Abdulrazaq, B.H. Chemical Peeling in the Treatment of Xeroderma Pigmentosum. Iraqi Prostgrad. Med. J. 2013, 12, 732–738. [Google Scholar]
- Wee, S.Y.; Ahn, D.-S. Facial resurfacing in xeroderma pigmentosum with chemical peeling. Plast. Reconstr. Surg. 1999, 103, 1464–1467. [Google Scholar] [CrossRef]
- Dayan, E.; Rohrich, R.J. Jessner’s Solution with Trichloroacetic Acid Chemical Peel: Optimizing Outcomes and Safety. Plast. Reconstr. Surg. Glob. Open 2019, 7, e2250. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.R.; Fader, D.J.; Gillard, M.; Baker, S.R.; Johnson, T.M.; Arbor, A. The role of dermabrasion and chemical peels in the treatment of patients with xeroderma pigmentosum. J. Am. Acad. Dermatol. 1995, 32, 623–626. [Google Scholar] [CrossRef]
- Bollag, W.; Ott, F. Retinoic acid: Topical treatment of senile or actinic keratoses and basal cell carcinomas. Agents Actions 1970, 1, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Philips, N.; Auler, S.; Hugo, R.; Gonzalez, S. Beneficial regulation of matrix metalloproteinases for skin health. Enzym. Res. 2011, 2011, 427285. [Google Scholar] [CrossRef] [PubMed]
- Kockaert, M.; Neumann, M. Systemic and topical drugs for aging skin. J. Drugs Dermatol. 2003, 2, 435–441. [Google Scholar]
- Ianhez, M.; Fleury Junior, L.F.F.; Miot, H.A.; Bagatin, E. Retinoids for prevention and treatment of actinic keratosis. An. Bras. Dermatol. 2013, 88, 585–593. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, Y.G.L.; Euvrard, S.; Bouwes Bavinck, J.N. Systemic and Topical Retinoids in the Management of Skin Cancer in Organ Transplant Recipients. Dermatol. Surg. 2004, 30, 656–661. [Google Scholar] [CrossRef]
- Bagatin, E.; Costa, C.S.; Rocha, M.A.D.D.; Picosse, F.R.; Kamamoto, C.S.L.; Pirmez, R.; Ianhez, M.; Miot, H.A. Consensus on the use of oral isotretinoin in dermatology-Brazilian Society of Dermatology. An. Bras. Dermatol. 2020, 95, 19–38. [Google Scholar] [CrossRef]
- DiGiovanna, J.J. Systemic retinoid therapy. Dermatol. Clin. 2001, 19, 161–167. [Google Scholar] [CrossRef]
- De Marchi, M.Â.; Maranhão, R.C.; Brandizzi, L.I.V.; Souza, D.R.S. Effects of isotretinoin on the metabolism of triglyceride-rich lipoproteins and on the lipid profile in patients with acne. Arch. Dermatol. Res. 2006, 297, 403–408. [Google Scholar] [CrossRef]
- Zeitouni, N.; Shieh, S.; Oseroff, A.R. Laser and Photodynamic Therapy in the Management of Cutaneous Malignancies. Clin. Dermatol. 2001, 19, 328–339. [Google Scholar] [CrossRef]
- Goncalves-Maia, M.; Magnaldo, T. Genetic therapy of Xeroderma Pigmentosum: Analysis of strategies and translation. Expert Opin. Orphan Drugs 2017, 5, 5–17. [Google Scholar] [CrossRef]
- Procianoy, F.; Cruz, A.A.V.; Baccega, A.; Ferraz, V.; Chahud, F. Aggravation of Eyelid and Conjunctival Malignancies Following Photodynamic Therapy in DeSanctis- Cacchione Syndrome. Ophthal. Plast. Reconstr. Surg. 2006, 22, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Cox, P.; Yarosh, D.B.; Kripke, M.L. Sunscreens and T4N5 liposomes differ in their ability to protect against ultraviolet-induced sunburn cell formation, alterations of dendritic epidermal cells, and local suppression of contact hypersensitivity. J. Investig. Dermatol. 1995, 104, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.M.; Cunningham, B.B. Photodynamic Therapy in a Teenage Girl with Xeroderma Pigmentosum Type C. Pediatr. Dermatol. 2012, 29, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Yang, Q.Q.; Ma, C.; Zou, D.X.; Wang, Y.X.; Sun, P.; Ju, A.Q.; Fang, F.; Gong, S.; Liu, W. Photodynamic therapy in the treatment of xeroderma pigmentosum: A case report. Photodiagn. Photodyn. Ther. 2020, 30, 101761. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.J. Ablative Laser Resurfacing for Skin Rejuvenation. Available online: https://www.uptodate.com/contents/ablative-laser-resurfacing-for-skin-rejuvenation (accessed on 10 September 2021).
- Hruza, G.J. Laser treatment of epidermal and dermal lesions. Dermatol. Clin. 2002, 20, 147–164. [Google Scholar] [CrossRef]
- Omi, T.; Numano, K. The Role of the CO2 Laser and Fractional CO2 Laser in Dermatology. Laser Ther. 2014, 23, 49–60. [Google Scholar] [CrossRef]
- Abdul-Wahid, S.N.; Shabba, K.; Ghaly Yousif, B.; Arowa, A.; Yousif, N.G. Use of laser in treatment of premalignant skin conditions. Pak. J. Biotechnol. 2018, 15, 763–767. [Google Scholar]
- Alster, T.; Lupton, J. Erbium:YAG cutaneous laser resurfacing. Dermatol. Clin. 2001, 19, 453–466. [Google Scholar] [CrossRef]
- Ross, E.V.; McKinlay, J.R.; Anderson, R.R. Why does carbon dioxide resurfacing work? A review. Arch. Dermatol. 1999, 135, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Friedli, A.; Bowes, L.; Kricorian, G.; Fitzpatrick, R.E. Full Face Laser Resurfacing: Therapy and Prophylaxis for Actinic Keratoses and Non-Melanoma Skin Cancer. Lasers Surg. Med. 2004, 34, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, J.U.; Quaedvlieg, P.J.F.; Neumann, M.H.A.M.; Krekels, G.A. Recurrence rates and long-term follow-up after laser resurfacing as a treatment for widespread actinic keratoses in the face and on the scalp. Dermatol. Surg. 2006, 32, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Rivers, J.K.; Rosoph, L.; Provost, N.; Bissonnette, R. Open-label study to assess the safety and efficacy of imiquimod 5% cream applied once daily three times per week in cycles for treatment of actinic keratoses on the head. J. Cutan. Med. Surg. 2008, 12, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Dinehart, S.; Whiting, D.; Lee, P.K.; Tawfik, N.; Jorizzo, J.; Lee, J.H.; Fox, T.L. Imiquimod 5% cream for the treatment of actinic keratosis: Results from two phase III, randomized, double-blind, parallel group, vehicle-controlled trials. J. Am. Acad. Dermatol. 2004, 50, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Korman, N.; Moy, R.; Ling, M.; Matheson, R.; Smith, S.; McKane, S.; Lee, J.H. Dosing With 5% Imiquimod Cream 3 Times per Week for the Treatment of Actinic Keratosis. Arch. Dermatol. 2005, 141, 467–473. [Google Scholar] [CrossRef]
- Szeimies, R.M.; Gerritsen, M.J.P.; Gupta, G.; Ortonne, J.P.; Serresi, S.; Bichel, J.; Lee, J.H.; Fox, T.L.; Alomar, A. Imiquimod 5% cream for the treatment of actinic keratosis: Results from a phase III, randomized, double-blind, vehicle-controlled, clinical trial with histology. J. Am. Acad. Dermatol. 2004, 51, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.K.; Harwell, W.B.; Loven, K.H.; Phillips, T.J.; Whiting, D.A.; Andres, K.L.; Lee, J.H. Long-term clinical outcomes following treatment of actinic keratosis with imiquimod 5% cream. Dermatol. Surg. 2005, 31, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Roseeuw, D. The treatment of basal skin carcinomas in two sisters with xeroderma pigmentosum. Clin. Exp. Dermatol. Suppl. 2003, 28, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.A.; Siegel, D.; Lyon, V.B.; Holland, K.E. Psoriasiform eruption and oral ulcerations as adverse effects of topical 5% imiquimod treatment in children: A report of four cases. Pediatr. Dermatol. 2013, 30, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Dika, E.; Neri, I.; Patrizi, A.; Lambertini, M.; Conti, F.; Pession, A.; Veronesi, G.; Scarfì, F.; Lacava, R. Imiquimod cream in pediatric patients: Recommendations, adverse events, and controversies. Dermatol. Ther. 2019, 32, e13116. [Google Scholar] [CrossRef] [PubMed]
- Carreau, M.; Quilliet, X.; Eveno, E.; Salvetti, A.; Danos, O.; Heard, J.M.; Mezzina, M.; Sarasin, A. Functional Retroviral Vector for Gene Therapy of Xeroderma Pigmentosum Group D Patients. Hum. Gene Ther. 1995, 1315, 1307–1315. [Google Scholar] [CrossRef]
- Zeng, L.; Quilliet, X.; Chevallier-Lagente, O.; Eveno, E.; Sarasin, A.; Mezzina, M. Retrovirus-mediated gene transfer corrects DNA repair defect of xeroderma pigmentosum cells of complementation groups A, B and C. Gene Ther. 1997, 4, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Sarasin, A.; Mezzina, M. Retrovirus-mediated DNA repair gene transfer into xeroderma pigmentosum cells: Perspectives for a gene therapy. Cell Biol. Toxicol. 1998, 14, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Fenjves, E.S.; Smith, J.; Zaradic, S.; Taichman, L.B. Systemic delivery of secreted protein by grafts of epidermal keratinocytes: Prospects for keratinocyte gene therapy. Hum. Gene Ther. 1994, 5, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Warrick, E.; Garcia, M.; Chagnoleau, C.; Chevallier, O.; Bergoglio, V.; Sartori, D.; Mavilio, F.; Angulo, J.F.; Avril, M.F.; Sarasin, A.; et al. Preclinical corrective gene transfer in xeroderma pigmentosum human skin stem cells. Mol. Ther. 2012, 20, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Menck, M.C.; Armelini, M.; Lima-Bessa, K. On the Search for Skin Gene Therapy Strategies of Xeroderma Pigmentosum Disease. Curr. Gene Ther. 2007, 7, 163–174. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dupuy, A.; Valton, J.; LeDuc, S.; Armier, J.; Galetto, R.; Gouble, A.; Lebuhotel, C.; Stary, A.; Pâques, F.; Duchateau, P.; et al. Targeted Gene Therapy of Xeroderma Pigmentosum Cells Using Meganuclease and Talen. PLoS ONE 2013, 8, 1–8. [Google Scholar] [CrossRef]
- Epinat, J.C. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003, 31, 2952–2962. [Google Scholar] [CrossRef]
- Sampson, T.R.; Weiss, D.S. Exploiting CRISPR/Cas systems for biotechnology. BioEssays 2014, 36, 34–38. [Google Scholar] [CrossRef]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef]
- Dupuy, A.; Sarasin, A. DNA damage and gene therapy of xeroderma pigmentosum, a human DNA repair-deficient disease. Mutat. Res. 2015, 776, 2–8. [Google Scholar] [CrossRef]
- Magnaldo, T.; Sarasin, A. Xeroderma pigmentosum: From Symptoms and Genetics to Gene-Based Skin Therapy. Cell Tissues Organs 2004, 177, 189–198. [Google Scholar] [CrossRef]
- Zahid, S.; Brownell, I. Repairing DNA damage in xeroderma pigmentosum: T4N5 lotion and gene therapy. J. Drugs Dermatol. 2008, 7, 405–408. [Google Scholar]
- Abu Lila, A.; Ishida, T. Liposomal Delivery Systems: Design Optimization and Current Applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Immordino, A.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Carita, A.C.; Eloy, J.O.; Chorilli, M.; Lee, R.J.; Leonardi, G.R. Recent Advances and Perspectives in Liposomes for Cutaneous Drug Delivery. Curr. Med. Chem. 2017, 25, 606–635. [Google Scholar] [CrossRef]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. [Google Scholar] [CrossRef]
- Patil, Y.; Amitay, Y.; Ohana, P.; Shmeeda, H.; Gabizon, A. Targeting of pegylated liposomal mitomycin-C prodrug to the folate receptor of cancer cells: Intracellular activation and enhanced cytotoxicity. J. Control. Release 2016, 225, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Crist, R.M.; Clogston, J.D.; McNeil, S.E. Zeta potential: A case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.; Roland, L.M.; Chu, L.F.; Campitelli, V.A.; Riley, E.T. Single-dose, extended-release epidural morphine (DepoDurTM) compared to conventional epidural morphine for post-Cesarean pain. Anesth. Analg. 2007, 105, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Usonis, V.; Bakasénas, V.; Valentelis, R.; Katiliene, G.; Vidzeniene, D.; Herzog, C. Antibody titres after primary and booster vaccination of infants and young children with a virosomal hepatitis A vaccine (Epaxal®). Vaccine 2003, 21, 4588–4592. [Google Scholar] [CrossRef]
- Mischler, R.; Metcalfe, I.C. Inflexal®V a trivalent virosome subunit influenza vaccine: Production. Vaccine 2002, 20, 5–11. [Google Scholar] [CrossRef]
- Lee, Y.; Thompson, D.H. Stimuli-responsive liposomes for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, 1–76. [Google Scholar] [CrossRef]
- Sugarman, S.M.; Perez-soler, R. Liposomes in the treatment of malignancy: A clinical perspective. Crit Rev. Oncol. Hematol. 1992, 12, 231–242. [Google Scholar] [CrossRef]
- Bardania, H.; Tarvirdipour, S.; Dorkoosh, F. Liposome-targeted delivery for highly potent drugs. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1478. [Google Scholar] [CrossRef]
- Lamb, Y.N. BNT162b2 mRNA COVID-19 Vaccine: First Approval. Drugs 2021, 81, 495–501. [Google Scholar] [CrossRef]
- Panahi, Y.; Farshbaf, M.; Mohammadhosseini, M.; Mirahadi, M.; Khalilov, R.; Saghfi, S.; Akbarzadeh, A. Recent advances on liposomal nanoparticles: Synthesis, characterization and biomedical applications. Artif. Cells Nanomed. Biotechnol. 2017, 45, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Bito, T.; Ueda, M.; Nagano, T.; Fujii, S.; Ichihashi, M. Reduction of ultraviolet-induced skin cancer in mice by topical application of DNA excision repair enzymes. Photodermatol. Photoimmunol. Photomed. 1995, 11, 9–13. [Google Scholar] [CrossRef]
- Dodson, M.L.; Lloyd, R.S. Structure-function studies of the T4 endonuclease V repair enzyme. Mutat. Res. Repair 1989, 218, 49–65. [Google Scholar] [CrossRef]
- Tanaka, K.; Hayakawa, H.; Sekiguchi, M.; Okada, Y. Specific action of T4 endonuclease V on damaged DNA in xeroderma pigmentosum cells in vivo. Proc. Natl. Acad. Sci. USA 1977, 74, 2958–2962. [Google Scholar] [CrossRef]
- Latham, K.A.; Taylor, J.S.; Lloyd, R.S. T4 endonuclease V protects the DNA strand opposite a thymine dimer from cleavage by the footprinting reagents DNase I and 1,10-phenanthroline-copper. J. Biol. Chem. 1995, 270, 3765–3771. [Google Scholar] [CrossRef]
- Tanaka, K. Restoration of ultraviolet-induced unscheduled DNA synthesis of xeroderma pigmentosum cells by the concomitant treatment with bacteriophage T4 endonuclease V and HVJ (Sendai virus). Proc. Natl. Acad. Sci. USA 1975, 72, 4071–4075. [Google Scholar] [CrossRef] [PubMed]
- Ceccoli, J.; Rosales, N.; Tsimis, J.; Yarosh, D. Encapsulation of the UV-DNA Repair Enzyme T4 Endonuclease V in Liposomes and Delivery to Human Cells. Soc. Investig. Dermatol. 1989, 93, 190–194. [Google Scholar] [CrossRef]
- Gilchrest, B.; Zhai, S.; Eller, M.; Yarosh, D.; Yaar, M. Treatment of human melanocytes and S91 melanoma cells with the DNA repair enzyme T4 endonuclease V enhances melanogenesis after ultraviolet irradiation. J. Investig. Dermatol. 1993, 101, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, D.; Kibitel, J.; Green, L.; Spinowitz, A. Enhanced unscheduled DNA synthesis in UV-irradiated human skin explants treated with T4N5 liposomes. J. Investig. Dermatol. 1991, 97, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, D.; Bucana, C.; Cox, P.; Alas, L.; Kibitel, J.; Kripke, M. Localization of liposomes containing a DNA repair enzyme in murine skin. J. Investig. Dermatol. 1994, 103, 461–468. [Google Scholar] [CrossRef]
- Kuchel, J.M.; St. Barnetson, R.C.; Halliday, G.M. Cyclobutane pyrimidine dimer formation is a molecular trigger for solar-simulated ultraviolet radiation-induced suppression of memory immunity in humans. Photochem. Photobiol. Sci. 2005, 4, 577–582. [Google Scholar] [CrossRef]
- Cafardi, J.A.; Elmets, C.A. T4 endonuclease V: Review and application to dermatology. Expert Opin. Biol. Ther. 2008, 8, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, D.B.; O’Connor, A.; Alas, L.; Potten, C.; Wolf, P. Photoprotection by Topical DNA Repair Enzymes: Molecular Correlates of Clinical Studies. Photochem. Photobiol. 1999, 69, 136–140. [Google Scholar] [CrossRef]
- Yarosh, D.; Klein, J.; O’Connor, A.; Hawk, J.; Rafal, E.; Wolf, P. Early report Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: A randomised study. Lancet 2001, 357, 926–929. [Google Scholar] [CrossRef]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Camp, W.L.; Turnham, J.W.; Athar, M.; Elmets, C.A. New Agents for Prevention of Ultraviolet-Induced Nonmelanoma Skin Cancer. Semin. Cutan. Med. Surg. 2011, 30, 6–13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lozzi, F.; Lanna, C.; Mazzeo, M.; Garofalo, V.; Palumbo, V.; Mazzilli, S.; Diluvio, L.; Terrinoni, A.; Bianchi, L.; Campione, E. Investigational drugs currently in phase II clinical trials for actinic keratosis. Expert Opin. Investig. Drugs 2019, 28, 629–642. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials. T4N5 Liposomal Lotion in Preventing the Recurrence of Nonmelanoma Skin Cancer in Patients Who Have Undergone a Kidney Transplant. 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT00089180?term=dimericine&draw=2&rank=1 (accessed on 10 September 2021).
- ClinicalTrials. T4N5 Liposome Lotion Compared With Placebo Lotion for Preventing Actinic Keratoses in Patients With Xeroderma Pigmentosum. 2013. Available online: https://clinicaltrials.gov/ct2/show/study/NCT00002811?term=dimericine&draw=2&rank=2 (accessed on 10 September 2021).
- De Andrade, F.A.G.; De Oliveira Cavalcanti, C.E.; Isoldi, F.C.; Ferreira, L.M. Therapeutics of xeroderma pigmentosum: A PRISMA-compliant systematic review. Indian J. Dermatol. Venereol. Leprol. 2021, 87, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Hacker, E.; Muller, H.; Hayward, N.; Fahey, P.; Walker, G. Enhancement of DNA repair using topical T4 endonuclease V does not inhibit melanoma formation in Cdk4(R24C/R24C)/Tyr-Nras(Q61K) mice following neonatal UVR. Pigment. Cell Melanoma Res. 2010, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Jefferies, C.; Cryan, S.-A. Targeted Liposomal Drug Delivery to Monocytes and Macrophages. J. Drug Deliv. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nagayasu, A.; Uchiyama, K.; Kiwada, H. The size of liposomes: A factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv. Drug Deliv. Rev. 1999, 40, 75–87. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccione, M.; Belloni Fortina, A.; Ferri, G.; Andolina, G.; Beretta, L.; Cividini, A.; De Marni, E.; Caroppo, F.; Citernesi, U.; Di Liddo, R. Xeroderma Pigmentosum: General Aspects and Management. J. Pers. Med. 2021, 11, 1146. https://doi.org/10.3390/jpm11111146
Piccione M, Belloni Fortina A, Ferri G, Andolina G, Beretta L, Cividini A, De Marni E, Caroppo F, Citernesi U, Di Liddo R. Xeroderma Pigmentosum: General Aspects and Management. Journal of Personalized Medicine. 2021; 11(11):1146. https://doi.org/10.3390/jpm11111146
Chicago/Turabian StylePiccione, Monica, Anna Belloni Fortina, Giulia Ferri, Gloria Andolina, Lorenzo Beretta, Andrea Cividini, Emanuele De Marni, Francesca Caroppo, Ugo Citernesi, and Rosa Di Liddo. 2021. "Xeroderma Pigmentosum: General Aspects and Management" Journal of Personalized Medicine 11, no. 11: 1146. https://doi.org/10.3390/jpm11111146
APA StylePiccione, M., Belloni Fortina, A., Ferri, G., Andolina, G., Beretta, L., Cividini, A., De Marni, E., Caroppo, F., Citernesi, U., & Di Liddo, R. (2021). Xeroderma Pigmentosum: General Aspects and Management. Journal of Personalized Medicine, 11(11), 1146. https://doi.org/10.3390/jpm11111146