Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein

 ,
,  ,
, 
Abstract

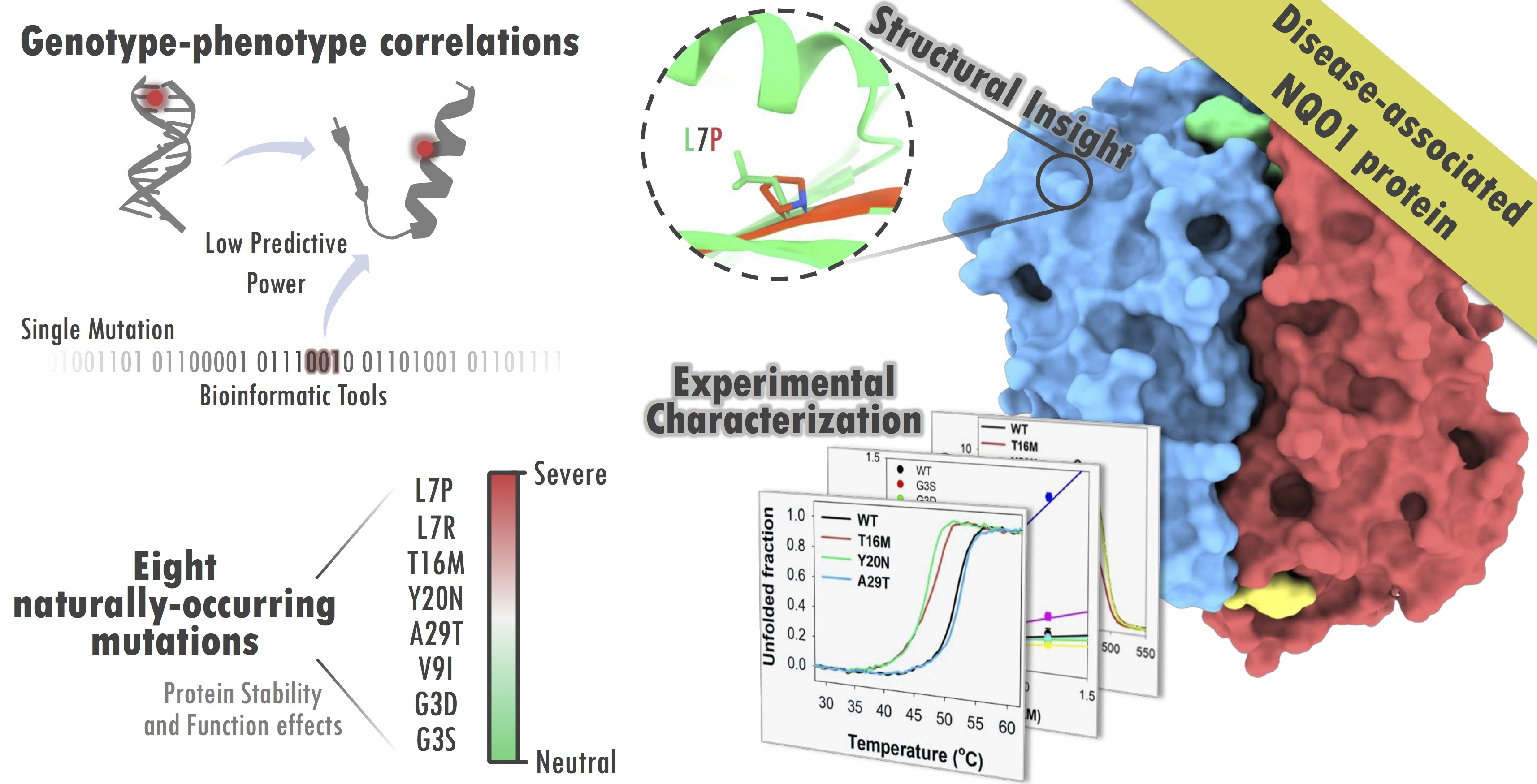
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. In Vitro Characterization of Purified Proteins
2.3. In Silico Mutagenesis and Structural Analysis
2.4. Structure- and Sequence-Based Analysis of Mutational Effects on Protein Stability and Potential Pathogenicity
3. Results and Discussion
3.1. Expression Analysis of NQO1 Variants Reveals Dramatic Effects of the Mutations L7R and L7P on Protein Stability and/or Solubility
3.2. Thermal Stability Analyses Revealed Significant Perturbation of the MMI by Mutations T16M and Y20N
3.3. The Local Stability of the Thermolysin Cleavage Site (TCS) Is Reduced by the Distant Mutations T16M and Y20N
3.4. The Mutations T16M, Y20N and A29T Perturb FAD Binding
3.5. None of the Mutations Affect Dic Binding Affinity or Energetics
3.6. Structural Analysis and Energy Calculations Provide Insight into Mutational Effects on Protein Stability and Function
3.6.1. The Mutations G3S and G3D
3.6.2. The Mutations L7P and L7R
3.6.3. The Mutation V9I
3.6.4. The Mutation T16M
3.6.5. The Mutation Y20N
3.6.6. The Mutation A29T
3.7. The Role of Protein Local Dynamics and Stability on Mutational Effects
3.8. Correlations between Loss-of-Function Scores Derived from Experimental Analysis of Mutational Effects and Bioinformatic Tools
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shendure, J.; Akey, J.M. The origins, determinants, and consequences of human mutations. Science 2015, 349, 1478–1483. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Goldstein, D.B.; Rowe, J.W.; Gharavi, A.G.; Wapner, R.; Chung, W.K. Precision Medicine in Internal Medicine. Ann. Intern. Med. 2019, 170, 635–642. [Google Scholar] [CrossRef]
- Stein, A.; Fowler, D.M.; Hartmann-Petersen, R.; Lindorff-Larsen, K. Biophysical and Mechanistic Models for Disease-Causing Protein Variants. Trends Biochem. Sci. 2019, 44, 475–488. [Google Scholar] [CrossRef]
- Manolio, T.A.; Fowler, D.M.; Starita, L.M.; Haendel, M.A.; MacArthur, D.G.; Biesecker, L.G.; Worthey, E.; Chisholm, R.L.; Green, E.D.; Jacob, H.J.; et al. Bedside Back to Bench: Building Bridges between Basic and Clinical Genomic Research. Cell 2017, 169, 6–12. [Google Scholar] [CrossRef]
- Casanueva, M.O.; Burga, A.; Lehner, B. Fitness trade-offs and environmentally induced mutation buffering in isogenic C. elegans. Science 2012, 335, 82–85. [Google Scholar] [CrossRef]
- Pey, A.L.; Stricher, F.; Serrano, L.; Martinez, A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am. J. Hum. Genet. 2007, 81, 1006–1024. [Google Scholar] [CrossRef]
- Nielsen, S.V.; Stein, A.; Dinitzen, A.B.; Papaleo, E.; Tatham, M.H.; Poulsen, E.G.; Kassem, M.M.; Rasmussen, L.J.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Predicting the impact of Lynch syndrome-causing missense mutations from structural calculations. PLoS Genet. 2017, 13, e1006739. [Google Scholar] [CrossRef]
- Abildgaard, A.B.; Stein, A.; Nielsen, S.V.; Schultz-Knudsen, K.; Papaleo, E.; Shrikhande, A.; Hoffmann, E.R.; Bernstein, I.; Gerdes, A.M.; Takahashi, M.; et al. Computational and cellular studies reveal structural destabilization and degradation of MLH1 variants in Lynch syndrome. eLife 2019, 8, e49138. [Google Scholar] [CrossRef] [PubMed]
- Blouin, J.M.; Bernardo-Seisdedos, G.; Sasso, E.; Esteve, J.; Ged, C.; Lalanne, M.; Sanz-Parra, A.; Urquiza, P.; de Verneuil, H.; Millet, O.; et al. Missense UROS mutations causing congenital erythropoietic porphyria reduce UROS homeostasis that can be rescued by proteasome inhibition. Hum. Mol. Genet. 2017, 26, 1565–1576. [Google Scholar] [CrossRef]
- Boycott, K.M.; Rath, A.; Chong, J.X.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; den Dunnen, J.T.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef]
- Mesa-Torres, N.; Fabelo-Rosa, I.; Riverol, D.; Yunta, C.; Albert, A.; Salido, E.; Pey, A.L. The role of protein denaturation energetics and molecular chaperones in the aggregation and mistargeting of mutants causing primary hyperoxaluria type I. PLoS ONE 2013, 8, e71963. [Google Scholar] [CrossRef]
- Pey, A.L.; Mesa-Torres, N.; Chiarelli, L.R.; Valentini, G. Structural and Energetic Basis of Protein Kinetic Destabilization in Human Phosphoglycerate Kinase 1 Deficiency. Biochemistry 2013, 52, 1160–1170. [Google Scholar] [CrossRef]
- Pey, A.L.; Maggi, M.; Valentini, G. Insights into human phosphoglycerate kinase 1 deficiency as a conformational disease from biochemical, biophysical, and in vitro expression analyses. J. Inherit. Metab. Dis. 2014, 37, 909–916. [Google Scholar] [CrossRef]
- Pey, A.L.; Padin-Gonzalez, E.; Mesa-Torres, N.; Timson, D.J. The metastability of human UDP-galactose 4′-epimerase (GALE) is increased by variants associated with type III galactosemia but decreased by substrate and cofactor binding. Arch. Biochem. Biophys. 2014, 562, 103–114. [Google Scholar] [CrossRef]
- Majtan, T.; Pey, A.L.; Gimenez-Mascarell, P.; Martinez-Cruz, L.A.; Szabo, C.; Kozich, V.; Kraus, J.P. Potential Pharmacological Chaperones for Cystathionine Beta-Synthase-Deficient Homocystinuria. Handb. Exp. Pharmacol. 2018, 245, 345–383. [Google Scholar]
- Fernandez-Higuero, J.A.; Betancor-Fernandez, I.; Mesa-Torres, N.; Muga, A.; Salido, E.; Pey, A.L. Structural and functional insights on the roles of molecular chaperones in the mistargeting and aggregation phenotypes associated with primary hyperoxaluria type I. Adv. Protein Chem. Struct. Biol. 2019, 114, 119–152. [Google Scholar]
- Medina-Carmona, E.; Betancor-Fernández, I.; Santos, J.; Mesa-Torres, N.; Grottelli, S.; Batlle, C.; Naganathan, A.N.; Oppici, O.; Cellini, B.; Ventura, S.; et al. Insight into the specificity and severity of pathogenic mechanisms associated with missense mutations through experimental and structural perturbation analyses. Hum. Mol. Genet. 2019, 28, 1–15. [Google Scholar] [CrossRef]
- Macias, I.; Lain, A.; Bernardo-Seisdedos, G.; Gil, D.; Gonzalez, E.; Falcon-Perez, J.M.; Millet, O. Hereditary tyrosinemia type I-associated mutations in fumarylacetoacetate hydrolase reduce the enzyme stability and increase its aggregation rate. J. Biol. Chem. 2019, 294, 13051–13060. [Google Scholar] [CrossRef] [PubMed]
- Fossbakk, A.; Kleppe, R.; Knappskog, P.M.; Martinez, A.; Haavik, J. Functional studies of tyrosine hydroxylase missense variants reveal distinct patterns of molecular defects in Dopa-responsive dystonia. Hum. Mutat. 2014, 35, 880–890. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Gleason, T.J.; Fridovich-Keil, J.L.; Timson, D.J. Misfolding of galactose 1-phosphate uridylyltransferase can result in type I galactosemia. Biochim. Biophys. Acta 2013, 1832, 1279–1293. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Kopec, J.; Pey, A.L.; Fitzpatrick, F.; Patel, D.; Chalk, R.; Streetha, L.; Yue, W.W. Molecular basis of classic galactosemia from the structure of human galactose 1-phosphate uridylyltransferase. Hum. Mol. Genet. 2016, 25, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Torres, N.; Betancor-Fernández, I.; Oppici, E.; Cellini, B.; Salido, E.; Pey, A.L. Evolutionary Divergent Suppressor Mutations in Conformational Diseases. Genes 2018, 9, 352. [Google Scholar] [CrossRef]
- Clausen, L.; Abildgaard, A.B.; Gersing, S.K.; Stein, A.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Protein stability and degradation in health and disease. Adv. Protein Chem. Struct. Biol. 2019, 114, 61–83. [Google Scholar] [PubMed]
- Scheller, R.; Stein, A.; Nielsen, S.V.; Marin, F.I.; Gerdes, A.M.; Marco, M.D.; Papaleo, E.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Towards mechanistic models for genotype-phenotype correlations in phenylketonuria using protein stability calculations. Hum. Mutat. 2019, 40, 444–457. [Google Scholar] [CrossRef]
- Erlandsen, H.; Pey, A.L.; Gamez, A.; Perez, B.; Desviat, L.R.; Aguado, C.; Koch, R.; Surendran, S.; Tyring, S.; Matalon, R.; et al. Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc. Natl. Acad. Sci. USA 2004, 101, 16903–16908. [Google Scholar] [CrossRef]
- Pey, A.L.; Majtan, T.; Sanchez-Ruiz, J.M.; Kraus, J.P. Human cystathionine beta-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): Complex regulation of CBS activity and stability by SAM. Biochem. J. 2013, 449, 109–121. [Google Scholar] [CrossRef]
- Fortian, A.; Castano, D.; Ortega, G.; Lain, A.; Pons, M.; Millet, O. Uroporphyrinogen III synthase mutations related to congenital erythropoietic porphyria identify a key helix for protein stability. Biochemistry 2009, 48, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Schmiesing, J.; Lohmoller, B.; Schweizer, M.; Tidow, H.; Gersting, S.W.; Muntau, A.C.; Braulke, T.; Muhlhausen, C. Disease-causing mutations affecting surface residues of mitochondrial glutaryl-CoA dehydrogenase impair stability, heteromeric complex formation and mitochondria architecture. Hum. Mol. Genet. 2017, 26, 538–551. [Google Scholar] [CrossRef][Green Version]
- Naganathan, A.N. Modulation of allosteric coupling by mutations: From protein dynamics and packing to altered native ensembles and function. Curr. Opin. Struct. Biol. 2018, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Sanchez, I.E.; Tejero, J.; Gomez-Moreno, C.; Medina, M.; Serrano, L. Point mutations in protein globular domains: Contributions from function, stability and misfolding. J. Mol. Biol. 2006, 363, 422–432. [Google Scholar] [CrossRef]
- Tokuriki, N.; Stricher, F.; Schymkowitz, J.; Serrano, L.; Tawfik, D.S. The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 2007, 369, 1318–1332. [Google Scholar] [CrossRef] [PubMed]
- Nisthal, A.; Wang, C.Y.; Ary, M.L.; Mayo, S.L. Protein stability engineering insights revealed by domain-wide comprehensive mutagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16367–16377. [Google Scholar] [CrossRef]
- Khan, S.; Vihinen, M. Performance of protein stability predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef]
- Roscoe, B.P.; Thayer, K.M.; Zeldovich, K.B.; Fushman, D.; Bolon, D.N. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J. Mol. Biol. 2013, 425, 1363–1377. [Google Scholar] [CrossRef]
- Pey, A.L. Biophysical and functional perturbation analyses at cancer-associated P187 and K240 sites of the multifunctional NADP(H):quinone oxidoreductase 1. Int. J. Biol. Macromol. 2018, 118, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta 2014, 1842, 2163–2173. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Palomino-Morales, R.J.; Fuchs, J.E.; Padín-Gonzalez, E.; Mesa-Torres, N.; Salido, E.; Timson, D.J.; Pey, A.L. Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep. 2016, 6, 20331. [Google Scholar] [CrossRef] [PubMed]
- Claveria-Gimeno, R.; Velazquez-Campoy, A.; Pey, A.L. Thermodynamics of cooperative binding of FAD to human NQO1: Implications to understanding cofactor-dependent function and stability of the flavoproteome. Arch Biochem. Biophys. 2017, 636, 17–27. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.J.; Pey, A.L. Enhanced vulnerability of human proteins towards disease-associated inactivation through divergent evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Neira, J.L.; Salido, E.; Fuchs, J.E.; Palomino-Morales, R.; Timson, D.J.; Pey, A.L. Site-to-site interdomain communication may mediate different loss-of-function mechanisms in a cancer-associated NQO1 polymorphism. Sci. Rep. 2017, 7, 44352. [Google Scholar] [CrossRef]
- Munoz, I.G.; Morel, B.; Medina-Carmona, E.; Pey, A.L. A mechanism for cancer-associated inactivation of NQO1 due to P187S and its reactivation by the consensus mutation H80R. FEBS Lett. 2017, 591, 2826–2835. [Google Scholar] [CrossRef]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Medina-Carmona, E.; Timson, D.J. Natural Small Molecules as Stabilizers and Activators of Cancer-Associated NQO1 Polymorphisms. Curr. Drug Targets 2016, 17, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H:quinone oxidoreductase(1.). FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Deng, P.S.; Bailey, J.M.; Swiderek, K.M. A two-domain structure for the two subunits of NAD(P)H:quinone acceptor oxidoreductase. Protein Sci. 1994, 3, 51–57. [Google Scholar] [CrossRef]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef]
- Martinez-Limon, A.; Alriquet, M.; Lang, W.H.; Calloni, G.; Wittig, I.; Vabulas, R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. USA 2016, 113, 12156–12161. [Google Scholar] [CrossRef]
- Salido, E.; Timson, D.J.; Betancor-Fernández, I.; Palomino-Morales, R.; Pey, A.L. Targeting HIF-1alpha Function in Cancer through the Chaperone Action of NQO1. Preprints 2020, 2020030285. [Google Scholar]
- Anusevicius, Z.; Sarlauskas, J.; Cenas, N. Two-electron reduction of quinones by rat liver NAD(P)H:quinone oxidoreductase: Quantitative structure-activity relationships. Arch. Biochem. Biophys. 2002, 404, 254–262. [Google Scholar] [CrossRef]
- Landi, L.; Fiorentini, D.; Galli, M.C.; Segura-Aguilar, J.; Beyer, R.E. DT-Diaphorase maintains the reduced state of ubiquinones in lipid vesicles thereby promoting their antioxidant function. Free Radic. Biol. Med. 1997, 22, 329–335. [Google Scholar] [CrossRef]
- Siegel, D.; Bolton, E.M.; Burr, J.A.; Liebler, D.C.; Ross, D. The reduction of alpha-tocopherolquinone by human NAD(P)H: Quinone oxidoreductase: The role of alpha-tocopherolhydroquinone as a cellular antioxidant. Mol. Pharmacol. 1997, 52, 300–305. [Google Scholar] [CrossRef]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. NQO1 in protection against oxidative stress. Curr. Opin. Toxicol. 2018, 7, 67–72. [Google Scholar] [CrossRef]
- Anoz-Carbonell, E.; Timson, D.J.; Pey, A.L.; Medina, M. The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity. Antioxidants 2020, 9, 772. [Google Scholar] [CrossRef]
- Timson, D.J. Dicoumarol: A Drug which Hits at Least Two Very Different Targets in Vitamin K Metabolism. Curr. Drug Targets 2017, 18, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.A.; Zhao, H.; Faulder, P.F.; Frenkel, A.D.; Timson, D.J.; Siegel, D.; Ross, D.; Burke, T.R., Jr.; Stratford, I.J.; Bryce, R.A. Coumarin-based inhibitors of human NAD(P)H:quinone oxidoreductase-1. Identification, structure-activity, off-target effects and in vitro human pancreatic cancer toxicity. J. Med. Chem. 2007, 50, 6316–6325. [Google Scholar] [CrossRef]
- Cullen, J.J.; Hinkhouse, M.M.; Grady, M.; Gaut, A.W.; Liu, J.; Zhang, Y.P.; Weydert, C.J.; Domann, F.E.; Oberley, L.W. Dicumarol inhibition of NADPH:quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res. 2003, 63, 5513–5520. [Google Scholar]
- Lajin, B.; Alachkar, A. The NQO1 polymorphism C609T (Pro187Ser) and cancer susceptibility: A comprehensive meta-analysis. Br. J. Cancer 2013, 109, 1325–1337. [Google Scholar] [CrossRef]
- Traver, R.D.; Horikoshi, T.; Danenberg, K.D.; Stadlbauer, T.H.; Danenberg, P.V.; Ross, D.; Gibson, N.W. NAD(P)H:quinone oxidoreductase gene expression in human colon carcinoma cells: Characterization of a mutation which modulates DT-diaphorase activity and mitomycin sensitivity. Cancer Res. 1992, 52, 797–802. [Google Scholar]
- Traver, R.D.; Siegel, D.; Beall, H.D.; Phillips, R.M.; Gibson, N.W.; Franklin, W.A.; Ross, D. Characterization of a polymorphism in NAD(P)H: Quinone oxidoreductase (DT-diaphorase). Br. J. Cancer 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Siegel, D.; Anwar, A.; Winski, S.L.; Kepa, J.K.; Zolman, K.L.; Ross, D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol. Pharmacol. 2001, 59, 263–268. [Google Scholar] [CrossRef]
- Eguchi-Ishimae, M.; Eguchi, M.; Ishii, E.; Knight, D.; Sadakane, Y.; Isoyama, K.; Yabe, H.; Mizutani, S.; Greaves, M. The association of a distinctive allele of NAD(P)H:quinone oxidoreductase with pediatric acute lymphoblastic leukemias with MLL fusion genes in Japan. Haematologica 2005, 90, 1511–1515. [Google Scholar] [PubMed]
- Lienhart, W.D.; Strandback, E.; Gudipati, V.; Koch, K.; Binter, A.; Uhl, M.K.; Rantasa, D.M.; Bourgeois, B.; Madl, T.; Zangger, K.; et al. Catalytic competence, structure and stability of the cancer-associated R139W variant of the human NAD(P)H:quinone oxidoreductase 1 (NQO1). FEBS J. 2017, 284, 1233–1245. [Google Scholar] [CrossRef]
- Pan, S.S.; Forrest, G.L.; Akman, S.A.; Hu, L.T. NAD(P)H:quinone oxidoreductase expression and mitomycin C resistance developed by human colon cancer HCT 116 cells. Cancer Res. 1995, 55, 330–335. [Google Scholar] [PubMed]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef]
- Fuchs, J.E.; Muñoz, I.G.; Timson, D.J.; Pey, A.L. Experimental and computational evidence on conformational fluctuations as a source of catalytic defects in genetic diseases. RSC Adv. 2016, 6, 58604. [Google Scholar] [CrossRef]
- Robertson, A.D.; Murphy, K.P. Protein Structure and the Energetics of Protein Stability. Chem. Rev. 1997, 97, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011, 79, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montano, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef]
- Bianchet, M.A.; Faig, M.; Amzel, L.M. Structure and mechanism of NAD[P]H:quinone acceptor oxidoreductases (NQO). Methods Enzymol. 2004, 382, 144–174. [Google Scholar]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A web server for structure-based protein stability prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO--multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Urolagin, S.; Vihinen, M. PON-P2: Prediction method for fast and reliable identification of harmful variants. PLoS ONE 2015, 10, e0117380. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Vankova, P.; Salido, E.; Timson, D.J.; Man, P.; Pey, A.L. A dynamic core in human NQO1 controls the functional and stability effects of ligand binding and their communication across the enzyme dimer. Biomolecules 2019, 9, 728. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Rizzuti, B.; Martin-Escolano, R.; Pacheco-Garcia, J.L.; Mesa-Torres, N.; Neira, J.L.; Guzzi, R.; Pey, A.L. Phosphorylation compromises FAD binding and intracellular stability of wild-type and cancer-associated NQO1: Insights into flavo-proteome stability. Int. J. Biol. Macromol. 2019, 125, 1275–1288. [Google Scholar] [CrossRef]
- Megarity, C.F.; Timson, D.J. Cancer-associated variants of human NQO1: Impacts on inhibitor binding and cooperativity. Biosci. Rep. 2019, 39, BSR20191874. [Google Scholar] [CrossRef]
- Scott, K.A.; Barnes, J.; Whitehead, R.C.; Stratford, I.J.; Nolan, K.A. Inhibitors of NQO1: Identification of compounds more potent than dicoumarol without associated off-target effects. Biochem. Pharmacol. 2011, 81, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Betancor-Fernandez, I.; Timson, D.J.; Salido, E.; Pey, A.L. Natural (and Unnatural) Small Molecules as Pharmacological Chaperones and Inhibitors in Cancer. Handb. Exp. Pharmacol. 2018, 45, 345–383. [Google Scholar]
- Jacob, J.; Duclohier, H.; Cafiso, D.S. The role of proline and glycine in determining the backbone flexibility of a channel-forming peptide. Biophys. J. 1999, 76, 1367–1376. [Google Scholar] [CrossRef]
- Morgan, A.A.; Rubenstein, E. Proline: The distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 2013, 8, e53785. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.E.; Baase, W.A.; Zhang, X.J.; Heinz, D.W.; Blaber, M.; Baldwin, E.P.; Matthews, B.W. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 1992, 255, 178–183. [Google Scholar] [CrossRef]
- Xue, M.; Wakamoto, T.; Kejlberg, C.; Yoshimura, Y.; Nielsen, T.A.; Risor, M.W.; Sanggaard, K.W.; Kitahara, R.; Mulder, F.A.A. How internal cavities destabilize a protein. Proc. Natl. Acad. Sci. USA 2019, 116, 21031–21036. [Google Scholar] [CrossRef]
- Pey, A.L. Towards Accurate Genotype–Phenotype Correlations in the CYP2D6 Gene. J. Pers. Med. 2020, 10, 158. [Google Scholar] [CrossRef]
- Gersting, S.W.; Kemter, K.F.; Staudigl, M.; Messing, D.D.; Danecka, M.K.; Lagler, F.B.; Sommerhoff, C.P.; Roscher, A.A.; Muntau, A.C. Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. Am. J. Hum. Genet. 2008, 83, 5–17. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Naganathan, A.N. A Self-Consistent Structural Perturbation Approach for Determining the Magnitude and Extent of Allosteric Coupling in Proteins. Biochem. J. 2017, 474, 2379–2388. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Sekhar, A.; Naganathan, A.N. A Universal Pattern in the Percolation and Dissipation of Protein Structural Perturbations. J. Phys. Chem. Lett. 2017, 8, 4779–4784. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Suresh, S.; Gopi, S.; Raman, K.; Naganathan, A.N. A General Mechanism for the Propagation of Mutational Effects in Proteins. Biochemistry 2017, 56, 294–305. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trivial Name | Amino Acid Change | Nucleotide Change | COSMIC | gnomAD V.2.1.1 | ΔΔG (kcal·mol−1) 1 | ||
|---|---|---|---|---|---|---|---|
| All Samples (AS) | Non-Cancer (NC) | Ratio (AS:NC) | |||||
| G3S | p.G3S | c.7G > A | - | 1.2 × 10−5 | 1.3 × 10−5 | 0.9 | −1.5 ± 1.0 |
| G3D | p.G3D | c.8G > A | + | - | - | - | −1.4 ± 0.9 |
| L7P | p.L7P | c.20T > C | + | - | - | - | −4.3 ± 1.2 |
| L7R | p.L7R | c.20T > G | - | 7.8 × 10−5 | 7.5 × 10−5 | 1.0 | −2.1 ± 0.8 |
| V9I | p.V9I | c.25G > A | - | 3.9 × 10−5 | 4.1 × 10−5 | 0.9 | −0.4 ± 0.6 |
| T16M | p.T16M | c.47C > T | - | 2.8 × 10−5 | 2.6 × 10−5 | 1.1 | −0.1 ± 0.2 |
| Y20N | p.Y20N | c.58T > A | - | 2.1 × 10−5 | 1.9 × 10−5 | 1.1 | −1.1 ± 0.6 |
| A29T | p.A29T | c.85G > A | + | - | - | - | −1.3 ± 1.3 |
| G3S | G3D | L7P | L7R | V9I | T16M | Y20N | A29T | |
|---|---|---|---|---|---|---|---|---|
| NQO1holo | −5.0 | −11.6 | −29.4 | −14.1 | 1.9 | −0.8 | −8.1 | −10.9 |
| NQO1dic | −1.5 | −10.1 | −25.5 | −35.9 | 0.6 | 0.8 | −5.4 | 0.4 |
| Variant | Expression | Thermal Stability | Proteolysis | FAD Binding | Dic Binding | ES |
|---|---|---|---|---|---|---|
| G3S | +++ | +++ | +++ | +++ | +++ | 3 |
| G3D | +++ | +++ | +++ | +++ | +++ | 3 |
| L7P | + | 1 | ||||
| L7R | + | 1 | ||||
| V9I | +++ | +++ | +++ | +++ | +++ | 3 |
| T16M | +++ | ++ | ++ | + | +++ | 2.2 |
| Y20N | +++ | ++ | +++ | ++ | +++ | 2.6 |
| A29T | +++ | +++ | +++ | ++ | +++ | 2.8 |
| P187S | +++ | + | + | + | + | 1.4 |
| P187E | + | 1 | ||||
| P187R | + | 1 | ||||
| P187L | + | 1 | ||||
| P187A | +++ | ++ | + | ++ | ++ | 2 |
| P187G | ++ | + | ++ | +++ | ++ | 2 |
| P187T | +++ | ++ | + | ++ | + | 1.8 |
| K240Q | +++ | +++ | ++ | ++ | ++ | 2.4 |
| K240I | +++ | +++ | ++ | +++ | +++ | 2.8 |
| K240E | +++ | +++ | + | ++ | ++ | 2.2 |
| K240T | +++ | +++ | +++ | ++ | +++ | 2.8 |
| K240H | +++ | +++ | ++ | ++ | +++ | 2.6 |
| K240A | +++ | +++ | + | +++ | +++ | 2.6 |
| K240G | +++ | +++ | + | ++ | ++ | 2.2 |
| Variant | PolyPhen-2 | SIFT | Mutation Taster | Provean | PON-P2 | BS |
|---|---|---|---|---|---|---|
| G3S | +++ | +++ | +++ | +++ | +++ | 3 |
| G3D | +++ | +++ | +++ | +++ | ++ | 2.8 |
| L7P | + | + | + | + | + | 1 |
| L7R | + | + | + | + | + | 1 |
| V9I | +++ | ++ | + | +++ | ++ | 2.2 |
| T16M | + | + | + | +++ | + | 1.4 |
| Y20N | ++ | +++ | + | +++ | ++ | 2.2 |
| A29T | +++ | +++ | + | +++ | +++ | 2.6 |
| P187S | +++ | +++ | +++ | + | ++ | 2 |
| P187E | ++ | + | N.Det. * | + | + | 1.25 |
| P187R | + | + | + | + | + | 1 |
| P187L | + | + | + | + | + | 1 |
| P187A | ++ | +++ | + | + | ++ | 1.8 |
| P187G | ++ | + | N.Det. * | + | + | 1.25 |
| P187T | ++ | +++ | + | + | + | 1.6 |
| K240Q | + | + | + | +++ | ++ | 1.6 |
| K240I | + | + | + | + | ++ | 1.2 |
| K240E | + | + | + | +++ | ++ | 1.6 |
| K240T | + | + | + | +++ | ++ | 1.6 |
| K240H | + | + | N.Det. * | +++ | ++ | 1.75 |
| K240A | + | + | N.Det. * | +++ | ++ | 1.75 |
| K240G | + | + | N.Det. * | + | ++ | 1.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco-García, J.L.; Cano-Muñoz, M.; Sánchez-Ramos, I.; Salido, E.; Pey, A.L. Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein. J. Pers. Med. 2020, 10, 207. https://doi.org/10.3390/jpm10040207
Pacheco-García JL, Cano-Muñoz M, Sánchez-Ramos I, Salido E, Pey AL. Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein. Journal of Personalized Medicine. 2020; 10(4):207. https://doi.org/10.3390/jpm10040207
Chicago/Turabian StylePacheco-García, Juan Luis, Mario Cano-Muñoz, Isabel Sánchez-Ramos, Eduardo Salido, and Angel L. Pey. 2020. "Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein" Journal of Personalized Medicine 10, no. 4: 207. https://doi.org/10.3390/jpm10040207
APA StylePacheco-García, J. L., Cano-Muñoz, M., Sánchez-Ramos, I., Salido, E., & Pey, A. L. (2020). Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein. Journal of Personalized Medicine, 10(4), 207. https://doi.org/10.3390/jpm10040207