Genetic Ablation of MiR-22 Fosters Diet-Induced Obesity and NAFLD Development

 ,
,  ,
, 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Maintenance of Constitutive Mir-22 Knockout (miR-22KO) Mice
2.3. Proteomic Analysis
2.4. Seahorse Analysis
2.5. Statistical Analysis
3. Results
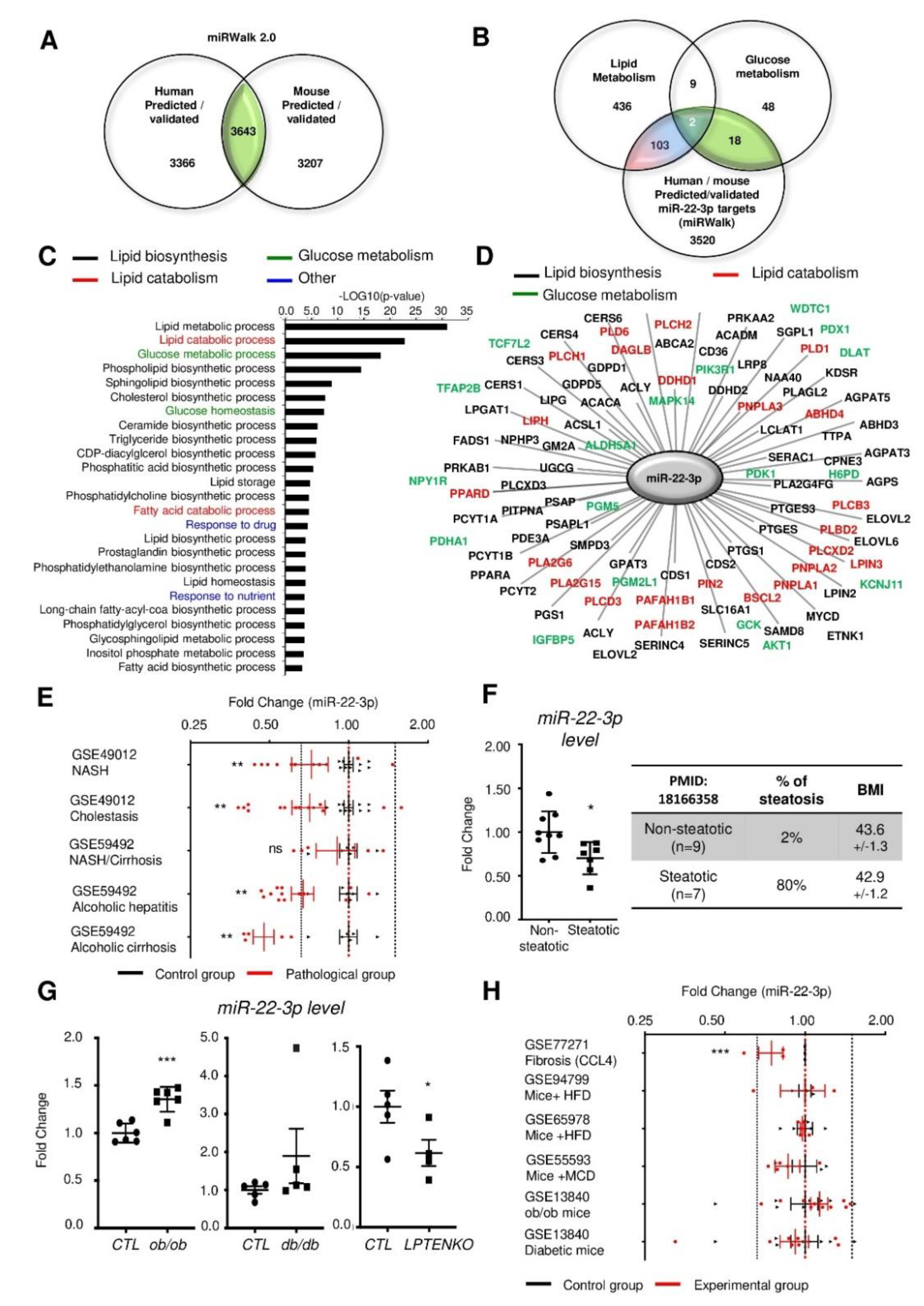
3.1. Mir-22-3p Expression in the Liver is Not a Reliable Marker of NAFLD and/or NASH.
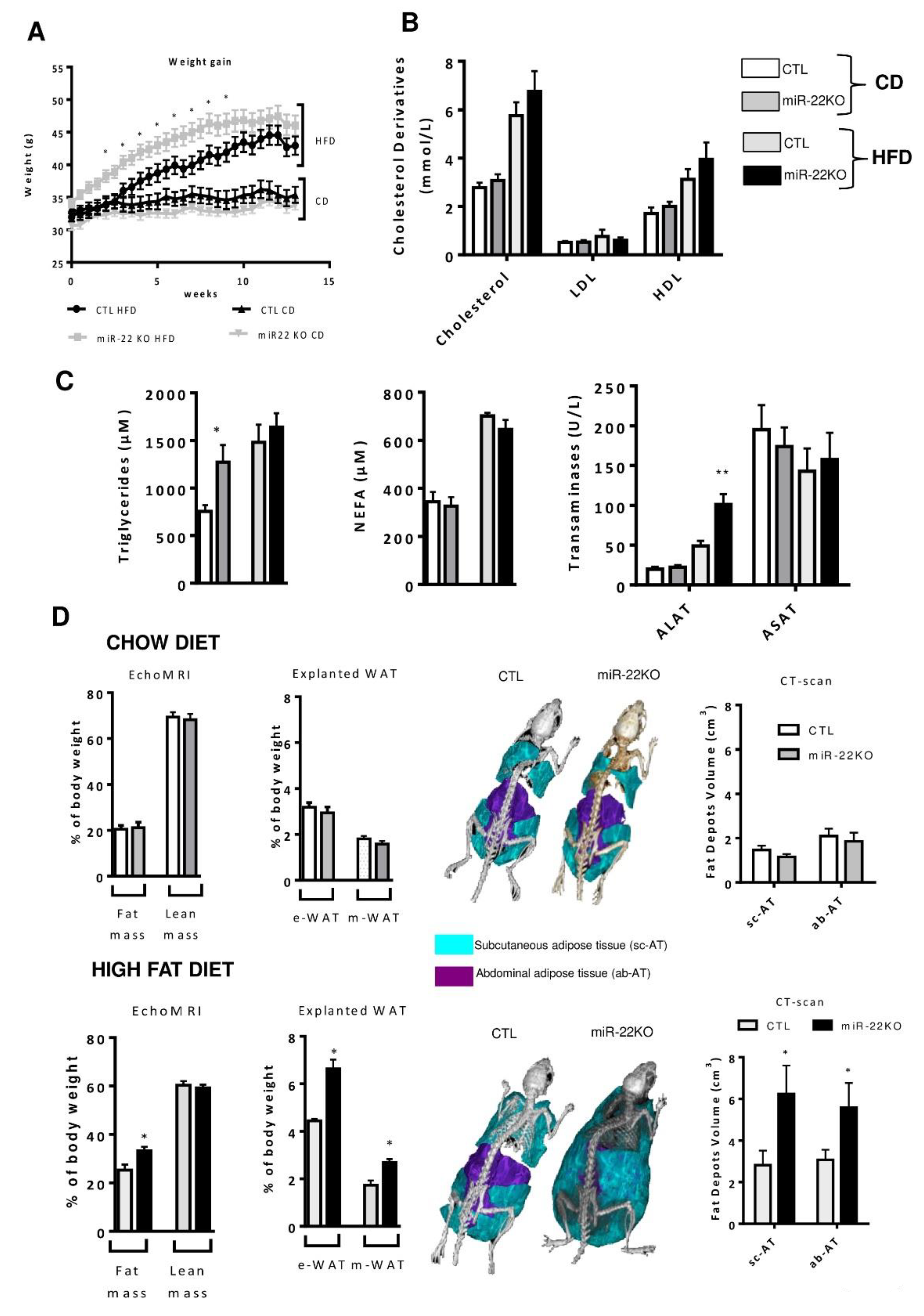
3.2. Mir22 Deletion Worsens Diet-Induced Obesity
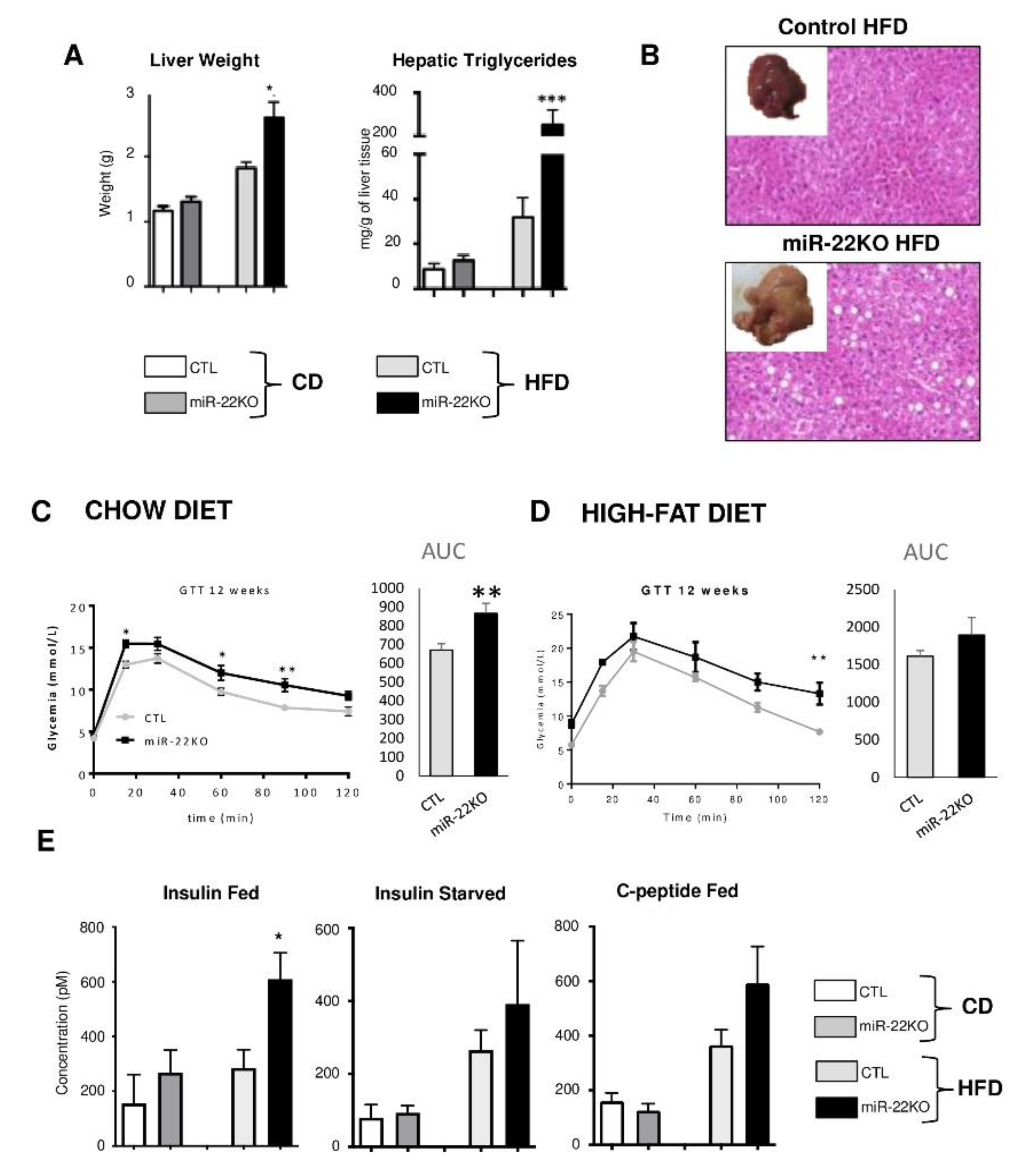
3.3. Mir22 Deletion in Mice Fosters Hepatic Steatosis and Glucose Intolerance
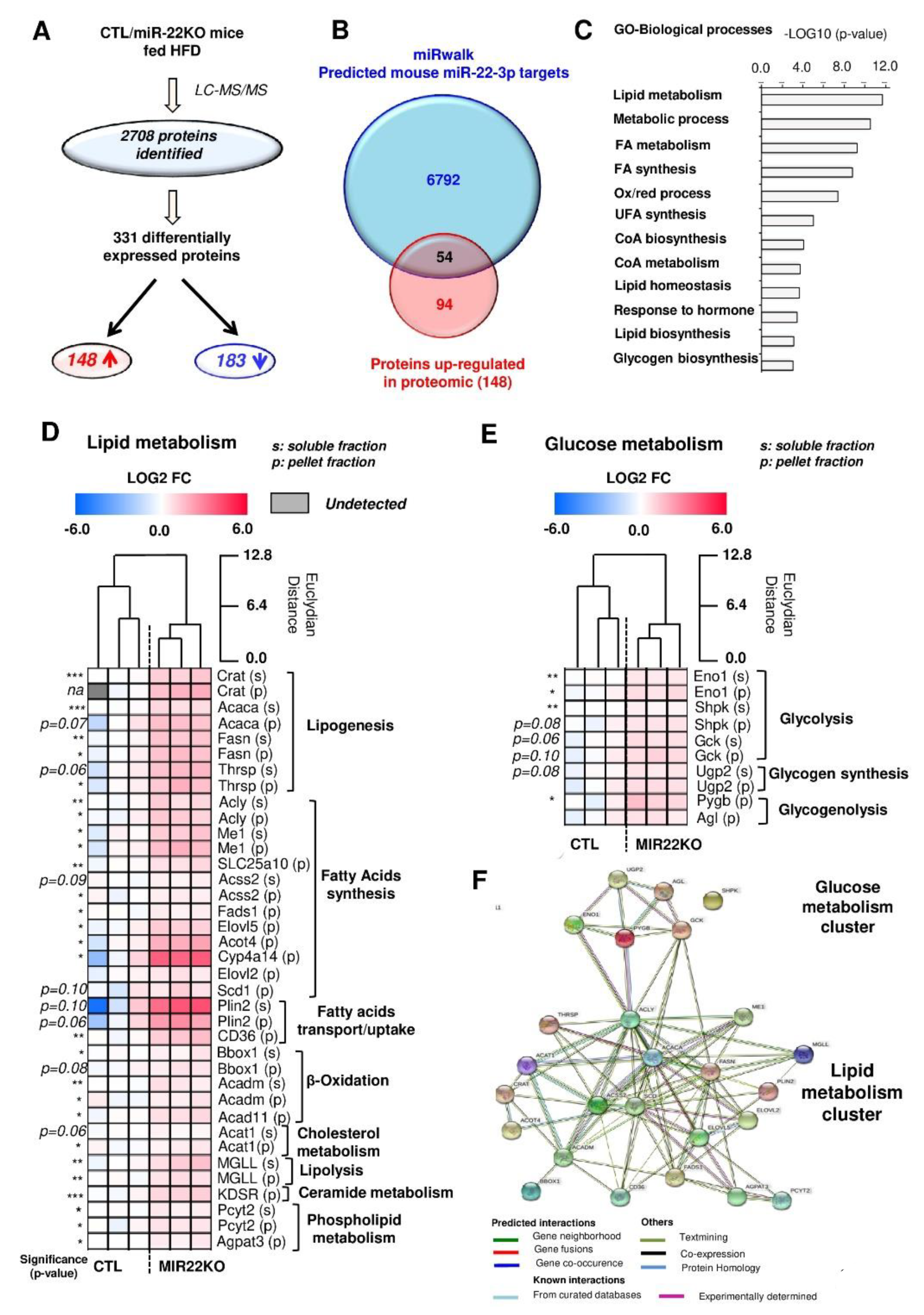
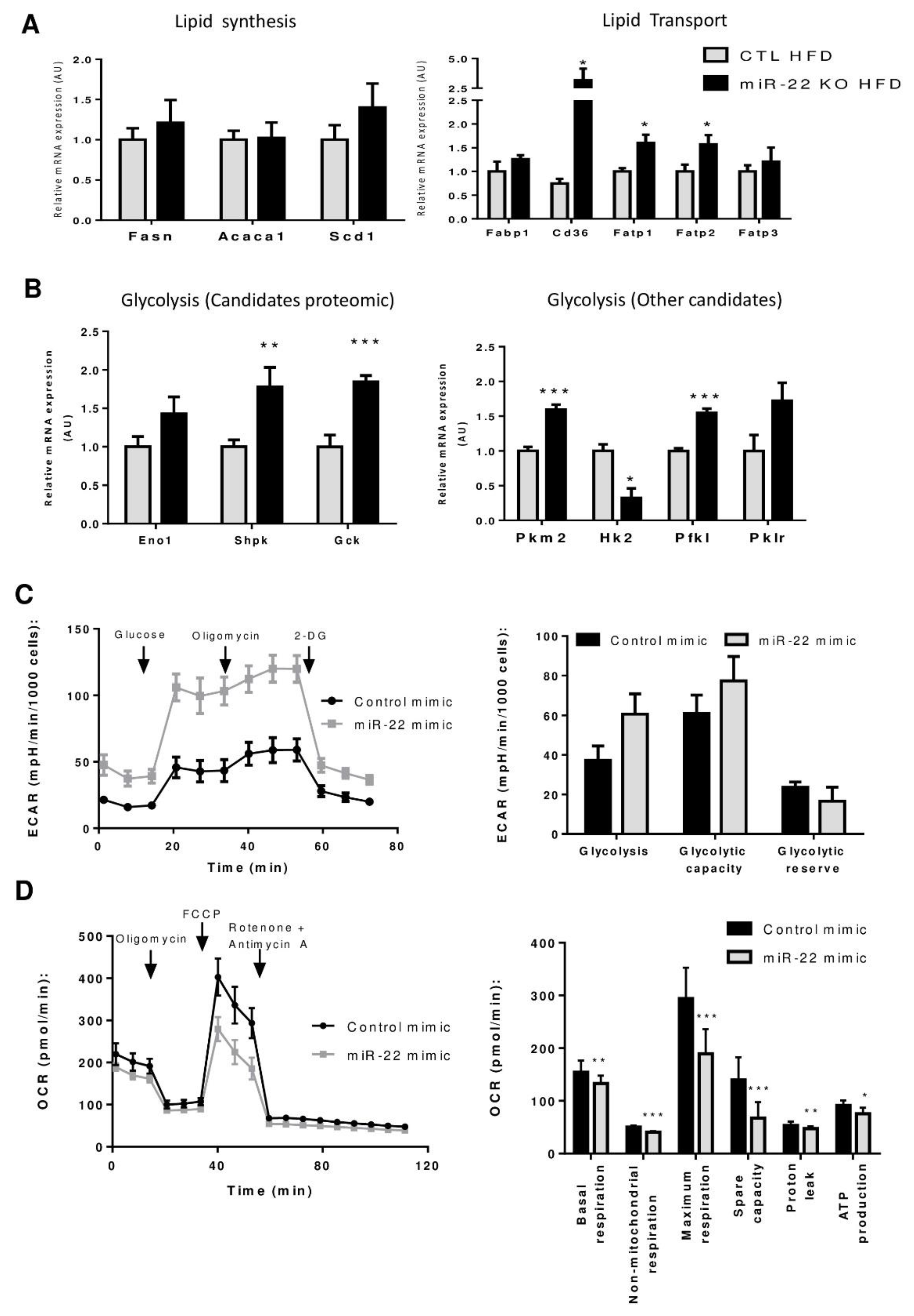
3.4. Proteomic Analysis of MiR-22KO Mice Liver Tissue Uncovers Alterations of Glycolysis and Hepatic Lipid Trafficking
3.5. Mir-22-3p Induces a Classic Warburg Effect in Hepatic Cancer Cell Line
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baffy, G. Hepatocellular Carcinoma in Non-alcoholic Fatty Liver Disease: Epidemiology, Pathogenesis, and Prevention. J. Clin. Transl. Hepatol. 2013, 1, 131–137. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Calo, N.; Portius, D.; Foti, M. MicroRNAs in fatty liver disease. Semin. Liver Dis. 2015, 35, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; De Sousa, M.C.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Ying, H.; Zhao, W.; Zhang, H. Analysis of microRNA expression profile induced by AICAR in mouse hepatocytes. Gene 2013, 512, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiao, Z.; Ji, J.; Li, S.; Huang, X.; Lu, X.; Zhao, H.; Peng, J.; Chen, X.; Ji, Q.; et al. Characterization of mouse serum exosomal small RNA content: The origins and their roles in modulating inflammatory response. Oncotarget 2017, 8, 42712–42727. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Vig, S.; Srivastava, R.; Mishra, A.; Singh, V.P.; Srivastava, A.K.; Datta, M. Elevated Hepatic miR-22-3p Expression Impairs Gluconeogenesis by Silencing the Wnt-Responsive Transcription Factor Tcf7. Diabetes 2015, 64, 3659–3669. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, H.-X.; Jena, P.K.; Sheng, L.; Ali, M.R.; Wan, Y.-J.Y. miR-22 inhibition reduces hepatic steatosis via FGF21 and FGFR1 induction. JHEP Rep. 2020, 2, 100093. [Google Scholar] [CrossRef]
- López-Riera, M.; Conde, I.; Quintas, G.; Pedrola, L.; Zaragoza, Á; Perez-Rojas, J.; Salcedo, M.; Benlloch, S.; Castell, J.V.; Jover, R. Non-invasive prediction of NAFLD severity: A comprehensive, independent validation of previously postulated serum microRNA biomarkers. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Senese, R.; Cioffi, F.; Petito, G.; De Lange, P.; Russo, A.; Goglia, F.; Lanni, A.; Potenza, N. miR-22-3p is involved in gluconeogenic pathway modulated by 3,5-diiodo-L-thyronine (T2). Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Diniz, G.P.; Huang, Z.-P.; Liu, J.; Chen, J.; Ding, J.; Fonseca, R.I.; Barreto-Chaves, M.L.M.; Donato, J.; Hu, X.; Wang, D.-Z. Loss of microRNA-22 prevents high-fat diet induced dyslipidemia and increases energy expenditure without affecting cardiac hypertrophy. Clin. Sci. 2017, 131, 2885–2900. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Lockstone, H.E.; Taylor, J.M.; Wills, Q.F.; Kaisaki, P.J.; Barrett, A.; Camps, C.; Fernandez, C.; Ragoussis, J.; Gauguier, D.; et al. MicroRNA-125a is over-expressed in insulin target tissues in a spontaneous rat model of Type 2 Diabetes. BMC Med. Genom. 2009, 2, 54. [Google Scholar] [CrossRef]
- Kaur, K.; Pandey, A.K.; Srivastava, S.; Srivastava, A.K.; Datta, M. Comprehensive miRNome and in silico analyses identify the Wnt signaling pathway to be altered in the diabetic liver. Mol. BioSyst. 2011, 7, 3234–3244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Yang, T.; Liu, Y.; Li, A.; Fu, S.; Wu, M.; Pan, Z.; Zhou, W. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br. J. Cancer 2010, 103, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, J.; Zhang, Y. MicroRNA-22 expression in hepatocellular carcinoma and its correlation with ezrin protein. J. Int. Med. Res. 2013, 41, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Zhang, L.-P.; Duan, C.-Y.; Wang, B.; He, N.-N.; Abulimiti, P.; Lin, Y. The inhibition role of miR-22 in hepatocellular carcinoma cell migration and invasion via targeting CD147. Cancer Cell Int. 2017, 17, 17. [Google Scholar] [CrossRef]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei-Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Haas, J.; Lieber, D.; Malterer, G.; Jaskiewicz, L.; Zavolan, M.; Dölken, L.; Zimmer, R. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014, 24, 906–919. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Veyrat–Durebex, C.; Moukil, M.A.; Rubbia–Brandt, L.; Rohner–Jeanrenaud, F.; Foti, M. PTEN down-regulation by unsaturated fatty acids triggers hepatic steatosis via an NF-kappaBp65/mTOR-dependent mechanism. Gastroenterology 2008, 134, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.-N.; Zhang, Y.-Q.; Wang, X.-M.; Liu, Y.-J.; Zhang, Y.-X.; Que, Y.-H.; Zhao, W.-J.; Li, P. Down-regulated miR-22 as predictive biomarkers for prognosis of epithelial ovarian cancer. Diagn. Pathol. 2014, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.-M.; Hu, L.-H.; Wang, Y.-Q.; Chen, P.; Huang, J.-G.; Lu, N.; He, J.-H.; Liao, C.-G. miR-22 is down-regulated in gastric cancer, and its overexpression inhibits cell migration and invasion via targeting transcription factor Sp1. Med. Oncol. 2013, 30, 542. [Google Scholar] [CrossRef]
- Calo, N.V.; Ramadori, P.; Sobolewski, C.; Romero, Y.; Maeder, C.; Fournier, M.; Rantakari, P.; Zhang, F.-P.; Poutanen, M.; Dufour, J.-F.J.; et al. Stress-activated miR-21/miR-21* in hepatocytes promotes lipid and glucose metabolic disorders associated with high-fat diet consumption. Gut 2016, 65, 1871–1881. [Google Scholar] [CrossRef]
- Chao, H.-W.; Chao, S.-W.; Lin, H.; Ku, H.-C.; Cheng, C.-F. Homeostasis of Glucose and Lipid in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 298. [Google Scholar] [CrossRef]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates with the Risk of Developing Liver Cancer. Front. Cell Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef]
- Alves, A.; Mamede, A.C.; Alves, M.G.; Oliveira, P.F.; Rocha, S.M.; Botelho, M.F.; Maia, C.J. Glycolysis Inhibition as a Strategy for Hepatocellular Carcinoma Treatment? Curr. Cancer Drug Targets 2019, 19, 26–40. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef]
- Morrisey, E.E. The magic and mystery of miR-21. J. Clin. Investig. 2010, 120, 3817–3819. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Wang, H.; Shi, J.; Wu, K.; Liu, S.; Liu, Y.; Wu, J. HCV-Induced miR-21 Contributes to Evasion of Host Immune System by Targeting MyD88 and IRAK1. PLoS Pathog. 2013, 9, e1003248. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Fiveash, C.E.; Braude, J.P.; Osborne, B.; Brown, S.H.; Mitchell, T.W.; Turner, N. Disparate metabolic response to fructose feeding between different mouse strains. Sci. Rep. 2015, 5, 18474. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Hallahan, N.L.; Brown, S.H.; Liu, M.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 2013, 56, 1129–1139. [Google Scholar] [CrossRef]
- Cruciani-Guglielmacci, C.; Bellini, L.; Denom, J.; Oshima, M.; Fernandez, N.; Normandie-Levi, P.; Berney, X.P.; Kassis, N.; Rouch, C.; Dairou, J.; et al. Molecular phenotyping of multiple mouse strains under metabolic challenge uncovers a role for Elovl2 in glucose-induced insulin secretion. Mol. Metab. 2017, 6, 340–351. [Google Scholar] [CrossRef]
- Harris, R.B.; Mitchell, T.D.; Yan, X.; Simpson, J.S.; Redmann, S.M. Metabolic responses to leptin in obese db/db mice are strain dependent. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R115–R132. [Google Scholar] [CrossRef] [PubMed]
- Madiehe, A.M.; Hebert, S.; Mitchell, T.D.; Harris, R.B.S. Strain-dependent stimulation of growth in leptin-treated obese db/db mice. Endocrinology 2002, 143, 3875–3883. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut 2016, 65, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Androsavich, J.R.; Chau, B.N.; Bhat, B.; Linsley, P.S.; Walter, N.G. Disease-linked microRNA-21 exhibits drastically reduced mRNA binding and silencing activity in healthy mouse liver. RNA 2012, 18, 1510–1526. [Google Scholar] [CrossRef] [PubMed]
- Luan, W.; Li, L.; Shi, Y.; Bu, X.; Xia, Y.; Wang, J.; Djangmah, H.S.; Liu, X.; You, Y.; Xu, B. Long non-coding RNA MALAT1 acts as a competing endogenous RNA to promote malignant melanoma growth and metastasis by sponging miR-22. Oncotarget 2016, 7, 63901–63912. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, W.; Jin, M.; Chen, J.; Xu, W.; Kong, X. lncRNA MIAT Functions as a Competing Endogenous RNA to Upregulate DAPK2 by Sponging miR-22-3p in Diabetic Cardiomyopathy. Cell Death Dis. 2017, 8, e2929. [Google Scholar] [CrossRef]
- Straniero, L.; Rimoldi, V.; Samarani, M.; Goldwurm, S.; Di Fonzo, A.; Krüger, R.; Deleidi, M.; Aureli, M.; Soldà, G.; Duga, S.; et al. The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Sci. Rep. 2017, 7, 12702. [Google Scholar] [CrossRef]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Zapalska-Sozoniuk, M.; Chrobak, L.; Kowalczyk, K.; Kankofer, M. Is it useful to use several “omics” for obtaining valuable results? Mol. Biol. Rep. 2019, 46, 3597–3606. [Google Scholar] [CrossRef]
- Luque-Garcia, J.L.; Zhou, G.; Spellman, D.S.; Sun, T.-T.; Neubert, T.A. Analysis of Electroblotted Proteins by Mass Spectrometry: Protein Identification after Western Blotting. Mol. Cell Proteom. 2008, 7, 308–314. [Google Scholar] [CrossRef]
- Qian, X.; Xu, W.; Xu, J.; Shi, Q.; Li, J.; Weng, Y.; Jiang, Z.; Feng, L.; Wang, X.; Zhou, J.; et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget 2017, 8, 47691–47708. [Google Scholar] [CrossRef]
- Mah, S.M.; Buske, C.; Humphries, R.K.; Kuchenbauer, F. miRNA*: A passenger stranded in RNA-induced silencing complex? Crit Rev. Eukaryot Gene Expr. 2010, 20, 141–148. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Pérez-Felpete, N.; Fernández-Fernández, C.; Donapetry-García, C.; Pazos-García, C. Liver glucose metabolism in humans. Biosci. Rep. 2016, 36, e00416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiang, D.; Hu, X.; Ruan, Q. Identification and study of differentially expressed miRNAs in aged NAFLD rats based on high-throughput sequencing. Ann. Hepatol. 2020, 19, 302–312. [Google Scholar] [CrossRef]
- Tian, H.; Zhu, X.; Lv, Y.; Jiao, Y.; Wang, G. Glucometabolic Reprogramming in the Hepatocellular Carcinoma Microenvironment: Cause and Effect. Cancer Manag. Res. 2020, 12, 5957–5974. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gjorgjieva, M.; Sobolewski, C.; Ay, A.-S.; Abegg, D.; Correia de Sousa, M.; Portius, D.; Berthou, F.; Fournier, M.; Maeder, C.; Rantakari, P.; et al. Genetic Ablation of MiR-22 Fosters Diet-Induced Obesity and NAFLD Development. J. Pers. Med. 2020, 10, 170. https://doi.org/10.3390/jpm10040170
Gjorgjieva M, Sobolewski C, Ay A-S, Abegg D, Correia de Sousa M, Portius D, Berthou F, Fournier M, Maeder C, Rantakari P, et al. Genetic Ablation of MiR-22 Fosters Diet-Induced Obesity and NAFLD Development. Journal of Personalized Medicine. 2020; 10(4):170. https://doi.org/10.3390/jpm10040170
Chicago/Turabian StyleGjorgjieva, Monika, Cyril Sobolewski, Anne-Sophie Ay, Daniel Abegg, Marta Correia de Sousa, Dorothea Portius, Flavien Berthou, Margot Fournier, Christine Maeder, Pia Rantakari, and et al. 2020. "Genetic Ablation of MiR-22 Fosters Diet-Induced Obesity and NAFLD Development" Journal of Personalized Medicine 10, no. 4: 170. https://doi.org/10.3390/jpm10040170
APA StyleGjorgjieva, M., Sobolewski, C., Ay, A.-S., Abegg, D., Correia de Sousa, M., Portius, D., Berthou, F., Fournier, M., Maeder, C., Rantakari, P., Zhang, F.-P., Poutanen, M., Picard, D., Montet, X., Nef, S., Adibekian, A., & Foti, M. (2020). Genetic Ablation of MiR-22 Fosters Diet-Induced Obesity and NAFLD Development. Journal of Personalized Medicine, 10(4), 170. https://doi.org/10.3390/jpm10040170