The Identification of Novel Biomarkers Is Required to Improve Adult SMA Patient Stratification, Diagnosis and Treatment


Abstract
1. Introduction
2. Evaluation of Functional Outcomes in Adult Patients
2.1. General Mobility Tests
2.2. Revised Upper Limb Module
2.3. Quantitative Assessment of Ambulation Capacity
2.4. Lung Function Tests
2.5. Additional Tests
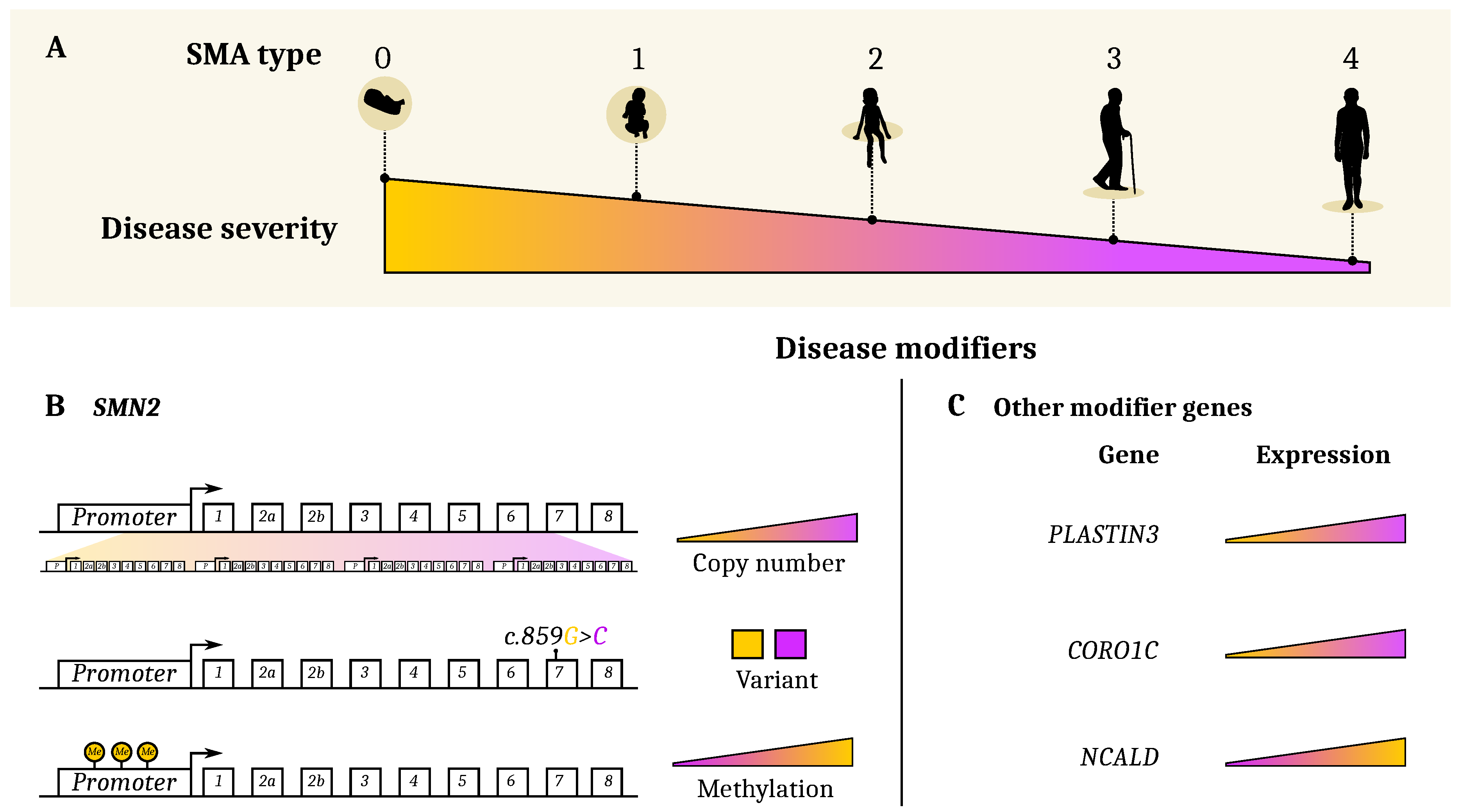
3. Genetic and Epigenetic Etiology of Clinical Heterogeneity
3.1. SMN2 and Other Genetic Modifiers
3.2. Plastin3
3.3. PLS3-Interacting Protein CORO1C and CHP1
3.4. NCALD
3.5. NAIP
3.6. Epigenetic Modifiers (Methylation)
3.7. Histone Deacetylases (HDAC) Inhibitors
4. Molecular Biomarkers
4.1. SMN Protein
4.2. Neurofilaments
4.3. Protein Tau
4.4. Serum Creatinine
5. Strategies for the Discoveries of Novel Biomarkers Towards the Development of Personalized Medicine Approaches for SMA
5.1. Multi-Omics Approaches for the Identification of SMA Biomarkers and Potential Therapeutic Targets
5.2. Perspectives for Personalized Medicine in Neuromuscular Disorders
6. Conclusions and Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Prior, T.W. Perspectives and diagnostic considerations in spinal muscular atrophy. Genet. Med. 2010, 12, 145–152. [Google Scholar] [CrossRef]
- Maretina, M.A.; Zheleznyakova, G.Y.; Lanko, K.M.; Egorova, A.A.; Baranov, V.S.; Kiselev, A.V. Molecular factors involved in spinal muscular atrophy pathways as possible disease-modifying candidates. Curr. Genom. 2018, 19, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008. [Google Scholar] [CrossRef]
- Cuscó, I.; Barceló, M.; Rojas-García, R.; Illa, I.; Gamez, J.; Cervera, C.; Pou, A.; Izquierdo, G.; Baiget, M.; Tizzano, E. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J. Neurol. 2006, 253, 21–25. [Google Scholar] [CrossRef]
- Jones, C.C.; Cook, S.F.; Jarecki, J.; Belter, L.; Reyna, S.P.; Staropoli, J.; Farwell, W.; Hobby, K. Spinal Muscular Atrophy (SMA) Subtype Concordance in Siblings: Findings From the Cure SMA Cohort. J. Neuromuscul. Dis. 2020, 7, 33–40. [Google Scholar] [CrossRef]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef]
- Oprea, G.E.; Kröber, S.; McWhorter, M.L.; Rossoll, W.; Müller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 2016, 99, 647–665. [Google Scholar] [CrossRef]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Löhr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Muela, N.; Litterman, N.K.; Norabuena, E.M.; Mull, J.L.; Galazo, M.J.; Sun, C.; Ng, S.Y.; Makhortova, N.R.; White, A.; Lynes, M.M.; et al. Single-cell analysis of SMN reveals its broader role in neuromuscular disease. Cell Rep. 2017, 18, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.Y.; Simpson, J.E.; Highley, J.R.; Heath, P.R. Spinal muscular atrophy: Factors that modulate motor neurone vulnerability. Neurobiol. Dis. 2017, 102, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; Darryl, C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Walter, M.C.; Wenninger, S.; Thiele, S.; Stauber, J.; Hiebeler, M.; Greckl, E.; Stahl, K.; Pechmann, A.; Lochmüller, H.; Kirschner, J.; et al. Safety and treatment effects of nusinersen in longstanding adult 5q-SMA type 3—A prospective observational study. J. Neuromuscul. Dis. 2019, 6, 453–465. [Google Scholar] [CrossRef]
- Hagenacker, T.; Wurster, C.D.; Günther, R.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Weiler, M.; Ziegler, A.; Kuttler, J.; Koch, J.C.; et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020, 19, 317–325. [Google Scholar] [CrossRef]
- Jochmann, E.; Steinbach, R.; Jochmann, T.; Chung, H.Y.; Rödiger, A.; Neumann, R.; Mayer, T.E.; Kirchhof, K.; Loudovici-Krug, D.; Smolenski, U.C.; et al. Experiences from treating seven adult 5q spinal muscular atrophy patients with Nusinersen. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420907803. [Google Scholar] [CrossRef] [PubMed]
- Deymeer, F.; Serdaroglu, P.; Parman, Y.; Poda, M. Natural history of SMA IIIb: Muscle strength decreases in a predictable sequence and magnitude. Neurology 2008, 71, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Tiziano, F.D.; Lomastro, R.; Di Pietro, L.; Pasanisi, M.B.; Fiori, S.; Angelozzi, C.; Abiusi, E.; Angelini, C.; Soraru, G.; Gaiani, A.; et al. Clinical and molecular cross-sectional study of a cohort of adult type III spinal muscular atrophy patients: Clues from a biomarker study. Eur. J. Hum. Genet. 2013, 21, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Kariyawasam, D.S.; D’silva, A.; Lin, C.; Ryan, M.M.; Farrar, M.A. Biomarkers and the development of a personalized medicine approach in spinal muscular atrophy. Front. Neurol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.; Wijngaarde, C.; Stam, M.; Bartels, B.; Otto, L.; Lemmink, H.; Schoenmakers, M.; Cuppen, I.; van den Berg, L.; van der Pol, W. Muscle strength and motor function throughout life in a cross-sectional cohort of 180 patients with spinal muscular atrophy types 1c–4. Eur. J. Neurol. 2018, 25, 512–518. [Google Scholar] [CrossRef]
- Wijngaarde, C.A.; Veldhoen, E.S.; van Eijk, R.P.; Stam, M.; Otto, L.A.; Asselman, F.L.; Wösten-van Asperen, R.M.; Hulzebos, E.H.; Verweij-van den Oudenrijn, L.P.; Bartels, B.; et al. Natural history of lung function in spinal muscular atrophy. Orphanet J. Rare Dis. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- O’Hagen, J.M.; Glanzman, A.M.; McDermott, M.P.; Ryan, P.A.; Flickinger, J.; Quigley, J.; Riley, S.; Sanborn, E.; Irvine, C.; Martens, W.B.; et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscul. Disord. 2007, 17, 693–697. [Google Scholar] [CrossRef]
- Kaufmann, P.; McDermott, M.P.; Darras, B.T.; Finkel, R.S.; Sproule, D.M.; Kang, P.B.; Oskoui, M.; Constantinescu, A.; Gooch, C.L.; Foley, A.R.; et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012, 79, 1889–1897. [Google Scholar] [CrossRef]
- Montes, J.; McDermott, M.P.; Mirek, E.; Mazzone, E.S.; Main, M.; Glanzman, A.M.; Duong, T.; Young, S.D.; Salazar, R.; Pasternak, A.; et al. Ambulatory function in spinal muscular atrophy: Age-related patterns of progression. PLoS ONE 2018, 13, e0199657. [Google Scholar] [CrossRef]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, N.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell. Mol. Med. 2020, 1–6. [Google Scholar] [CrossRef]
- Kessler, T.; Latzer, P.; Schmid, D.; Warnken, U.; Saffari, A.; Ziegler, A.; Kollmer, J.; Möhlenbruch, M.; Ulfert, C.; Herweh, C.; et al. Cerebrospinal fluid proteomic profiling in nusinersen-treated patients with spinal muscular atrophy. J. Neurochem. 2020, e14953. [Google Scholar] [CrossRef]
- Montes, J.; Glanzman, A.M.; Mazzone, E.S.; Martens, W.B.; Dunaway, S.; Pasternak, A.; Riley, S.O.; Quigley, J.; Pandya, S.; De Vivo, D.C.; et al. Spinal muscular atrophy functional composite score: A functional measure in spinal muscular atrophy. Muscle Nerve 2015, 52, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, E.; Bianco, F.; Martinelli, D.; Glanzman, A.M.; Messina, S.; De Sanctis, R.; Main, M.; Eagle, M.; Florence, J.; Krosschell, K.; et al. Assessing upper limb function in nonambulant SMA patients: Development of a new module. Neuromuscul. Disord. 2011, 21, 406–412. [Google Scholar] [CrossRef]
- Mazzone, E.S.; Mayhew, A.; Montes, J.; Ramsey, D.; Fanelli, L.; Young, S.D.; Salazar, R.; De Sanctis, R.; Pasternak, A.; Glanzman, A.; et al. Revised upper limb module for spinal muscular atrophy: Development of a new module. Muscle Nerve 2017, 55, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.C.; Coratti, G.; Mazzone, E.S.; Montes, J.; Scoto, M.; De Sanctis, R.; Main, M.; Mayhew, A.; Muni Lofra, R.; Dunaway Young, S.; et al. Revised upper limb module for spinal muscular atrophy: 12 month changes. Muscle Nerve 2019, 59, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Kizina, K.; Stolte, B.; Totzeck, A.; Bolz, S.; Fleischer, M.; Mönninghoff, C.; Guberina, N.; Oldenburg, D.; Forsting, M.; Kleinschnitz, C.; et al. Clinical implication of dosimetry of computed tomography-and fluoroscopy-guided intrathecal therapy with nusinersen in adult patients with spinal muscular atrophy. Front. Neurol. 2019, 10, 1166. [Google Scholar] [CrossRef]
- Stolte, B.; Totzeck, A.; Kizina, K.; Bolz, S.; Pietruck, L.; Mönninghoff, C.; Guberina, N.; Oldenburg, D.; Forsting, M.; Kleinschnitz, C.; et al. Feasibility and safety of intrathecal treatment with nusinersen in adult patients with spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418803246. [Google Scholar] [CrossRef]
- Zerres, K.; Rudnik-Schöneborn, S.; Forrest, E.; Lusakowska, A.; Borkowska, J.; Hausmanowa-Petrusewicz, I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J. Neurol. Sci. 1997, 146, 67–72. [Google Scholar] [CrossRef]
- ATS statement: Guidelines for the six-minute walk test. Am. J. Respir. Crit. Care Med. 2002, 166, 111–117. [CrossRef]
- Dunaway Young, S.; Montes, J.; Kramer, S.S.; Marra, J.; Salazar, R.; Cruz, R.; Chiriboga, C.A.; Garber, C.E.; De Vivo, D.C. Six-minute walk test is reliable and valid in spinal muscular atrophy. Muscle Nerve 2016, 54, 836–842. [Google Scholar] [CrossRef]
- McKay, M.J.; Baldwin, J.N.; Ferreira, P.; Simic, M.; Vanicek, N.; Burns, J.; For the 1000 Norms Project Consortium. Reference values for developing responsive functional outcome measures across the lifespan. Neurology 2017, 88, 1512–1519. [Google Scholar] [CrossRef]
- Mills, K.R. The basics of electromyography. J. Neurol. Neurosurg. Psychiatry 2005, 76, ii32–ii35. [Google Scholar] [CrossRef] [PubMed]
- Kimura, J. Peripheral nerve conduction studies and neuromuscular junction testing. In Handbook of Clinical Neurophysiology; Elsevier: Amsterdam, The Netherlands, 2004; Volume 4, pp. 241–270. [Google Scholar]
- Arnold, W.D.; Porensky, P.N.; McGovern, V.L.; Iyer, C.C.; Duque, S.; Li, X.; Meyer, K.; Schmelzer, L.; Kaspar, B.K.; Kolb, S.J.; et al. Electrophysiological biomarkers in spinal muscular atrophy: Proof of concept. Ann. Clin. Transl. Neurol. 2014, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Nandedkar, S.D.; Nandedkar, D.S.; Barkhaus, P.E.; Stalberg, E.V. Motor unit number index (MUNIX). IEEE Tans. Biomed. Eng. 2004, 51, 2209–2211. [Google Scholar] [CrossRef] [PubMed]
- Hausmanowa-Petrusewicz, I. Role of electromyography in the diagnosis of motor neuron disorders. Neuropatol. Pol. 1992, 30, 187–197. [Google Scholar]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T.; et al. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef]
- Swoboda, K.J.; Prior, T.W.; Scott, C.B.; McNaught, T.P.; Wride, M.C.; Reyna, S.P.; Bromberg, M.B. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann. Neurol. Offi. J. Am. Neurol. Assoc. Child Neurol. Soc. 2005, 57, 704–712. [Google Scholar] [CrossRef]
- Querin, G.; Lenglet, T.; Debs, R.; Stojkovic, T.; Béhin, A.; Salachas, F.; Le Forestier, N.; del Mar Amador, M.; Lacomblez, L.; Meininger, V.; et al. The motor unit number index (MUNIX) profile of patients with adult spinal muscular atrophy. Clini. Neurophysiol. 2018, 129, 2333–2340. [Google Scholar] [CrossRef]
- Querin, G.; El Mendili, M.M.; Lenglet, T.; Behin, A.; Stojkovic, T.; Salachas, F.; Devos, D.; Le Forestier, N.; del Mar Amador, M.; Debs, R.; et al. The spinal and cerebral profile of adult spinal-muscular atrophy: A multimodal imaging study. NeuroImage Clin. 2019, 21, 101618. [Google Scholar] [CrossRef]
- Taso, M.; Le Troter, A.; Sdika, M.; Ranjeva, J.P.; Guye, M.; Bernard, M.; Callot, V. Construction of an in vivo human spinal cord atlas based on high-resolution MR images at cervical and thoracic levels: Preliminary results. Magn. Reson. Mater. Phys. Biol. Med. 2014, 27, 257–267. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Scharf, J.M.; Endrizzi, M.G.; Wetter, A.; Huang, S.; Thompson, T.G.; Zerres, K.; Dietrich, W.F.; Wirth, B.; Kunkel, L.M. Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat. Genet. 1998, 20, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Talbot, K. Spinal muscular atrophies reveal motor neuron vulnerability to defects in ribonucleoprotein handling. Curr. Opin. Neurol. 2003, 16, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Bürglen, L.; Seroz, T.; Miniou, P.; Lefebvre, S.; Burlet, P.; Munnich, A.; Pequignot, E.V.; Egly, J.M.; Melki, J. The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am. J. Hum. Genet. 1997, 60, 72. [Google Scholar] [PubMed]
- Jiang, J.; Huang, J.; Gu, J.; Cai, X.; Zhao, H.; Lu, H. Genomic analysis of a spinal muscular atrophy (SMA) discordant family identifies a novel mutation in TLL2, an activator of growth differentiation factor 8 (myostatin): A case report. BMC Med. Genet. 2019, 20, 204. [Google Scholar] [CrossRef]
- Van der Steege, G.; Grootscholten, P.; Van der Vlies, P.; Draaijers, T.; Osinga, J.; Cobben, J.; Scheffer, H.; Buys, C. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995, 345, 985–986. [Google Scholar] [CrossRef]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997, 6, 1205–1214. [Google Scholar] [CrossRef]
- McAndrew, P.; Parsons, D.; Simard, L.; Rochette, C.; Ray, P.; Mendell, J.; Prior, T.; Burghes, A. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997, 60, 1411–1422. [Google Scholar] [CrossRef]
- Taylor, J.E.; Thomas, N.H.; Lewis, C.M.; Abbs, S.J.; Rodrigues, N.R.; Davies, K.E.; Mathew, C.G. Correlation of SMN t and SMN c gene copy number with age of onset and survival in spinal muscular atrophy. Eur. J. Hum. Genet. 1998, 6, 467–474. [Google Scholar] [CrossRef]
- Mailman, M.D.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Wirth, B.; Burghes, A.H.; Prior, T.W. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar] [CrossRef]
- Anhuf, D.; Eggermann, T.; Rudnik-Schöneborn, S.; Zerres, K. Determination of SMN1 and SMN2 copy number using TaqMan™ technology. Hum. Mutat. 2003, 22, 74–78. [Google Scholar] [CrossRef]
- Scarciolla, O.; Stuppia, L.; De Angelis, M.V.; Murru, S.; Palka, C.; Giuliani, R.; Pace, M.; Di Muzio, A.; Torrente, I.; Morella, A.; et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006, 7, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Brichta, L.; Schrank, B.; Lochmüller, H.; Blick, S.; Baasner, A.; Heller, R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet. 2006, 119, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Curet, I.; Robinson, K.G.; Funanage, V.L.; Crawford, T.O.; Scavina, M.; Wang, W. Robust quantification of the SMN gene copy number by real-time TaqMan PCR. Neurogenetics 2007, 8, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Chang, Y.Y.; Chen, C.H.; Kuo, Y.S.; Hwu, W.L.; Gerdes, T.; Ko, T.M. Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet. Med. 2007, 9, 241–248. [Google Scholar] [CrossRef]
- Tiziano, F.; Bertini, E.; Messina, S.; Angelozzi, C.; Pane, M.; D’Amico, A.; Alfieri, P.; Fiori, S.; Battini, R.; Berardinelli, A.; et al. The Hammersmith functional score correlates with the SMN2 copy number: A multicentric study. Neuromuscul. Disord. 2007, 17, 400–403. [Google Scholar] [CrossRef]
- Elsheikh, B.; Prior, T.; Zhang, X.; Miller, R.; Kolb, S.J.; Moore, D.; Bradley, W.; Barohn, R.; Bryan, W.; Gelinas, D.; et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2009, 40, 652–656. [Google Scholar] [CrossRef]
- Alias, L.; Bernal, S.; Barceló, M.J.; Also-Rallo, E.; Martínez-Hernández, R.; Rodriguez-Alvarez, F.J.; Hernández-Chico, C.; Baiget, M.; Tizzano, E.F. Accuracy of marker analysis, quantitative real-time polymerase chain reaction, and multiple ligation-dependent probe amplification to determine SMN2 copy number in patients with spinal muscular atrophy. Genet. Test. Mol. Biomark. 2011, 15, 587–594. [Google Scholar] [CrossRef]
- Amara, A.; Adala, L.; Charfeddine, I.B.; Mamaï, O.; Mili, A.; Lazreg, T.B.; H’mida, D.; Amri, F.; Salem, N.; Boughammura, L.; et al. Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur. J. Paediatric Neurol. 2012, 16, 167–174. [Google Scholar] [CrossRef]
- Crawford, T.O.; Paushkin, S.V.; Kobayashi, D.T.; Forrest, S.J.; Joyce, C.L.; Finkel, R.S.; Kaufmann, P.; Swoboda, K.J.; Tiziano, D.; Lomastro, R.; et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS ONE 2012, 7, e33572. [Google Scholar] [CrossRef]
- Dobrowolski, S.F.; Pham, H.T.; Pouch Downes, F.; Prior, T.W.; Naylor, E.W.; Swoboda, K.J. Newborn screening for spinal muscular atrophy by calibrated short-amplicon melt profiling. Clin. Chem. 2012, 58, 1033–1039. [Google Scholar] [CrossRef]
- Kirwin, S.M.; Vinette, K.M.; Gonzalez, I.L.; Abdulwahed, H.A.; Al-Sannaa, N.; Funanage, V.L. A homozygous double mutation in SMN 1: A complicated genetic diagnosis of SMA. Mol. Genet. Genom. Med. 2013, 1, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Brkušanin, M.; Kosać, A.; Jovanović, V.; Pešović, J.; Brajušković, G.; Dimitrijević, N.; Todorović, S.; Romac, S.; Rašić, V.M.; Savić-Pavićević, D. Joint effect of the SMN2 and SERF1A genes on childhood-onset types of spinal muscular atrophy in Serbian patients. J. Hum. Genet. 2015, 60, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Li, L.; Zeng, J.; Zhou, W.J.; Wu, W.Q.; Zhong, Z.Y.; Yan, T.Z.; Xie, J.S.; Huang, J.; Lin, L.; et al. Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy and unrelated healthy controls. BMC Musculoskel. Disord. 2015, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Stabley, D.L.; Harris, A.W.; Holbrook, J.; Chubbs, N.J.; Lozo, K.W.; Crawford, T.O.; Swoboda, K.J.; Funanage, V.L.; Wang, W.; Mackenzie, W.; et al. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol. Genet. Genom. Med. 2015, 3, 248–257. [Google Scholar] [CrossRef]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef]
- Bernal, S.; Alias, L.; Barcelo, M.; Also-Rallo, E.; Martínez-Hernández, R.; Gámez, J.; Guillén-Navarro, E.; Rosell, J.; Hernando, I.; Rodríguez-Alvarez, F.; et al. The c. 859G> C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J. Med. Genet. 2010, 47, 640–642. [Google Scholar] [CrossRef]
- Vezain, M.; Saugier-Veber, P.; Goina, E.; Touraine, R.; Manel, V.; Toutain, A.; Fehrenbach, S.; Frébourg, T.; Pagani, F.; Tosi, M.; et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010, 31, E1110–E1125. [Google Scholar] [CrossRef]
- Burghes, A.H.; Ingraham, S.E.; McLean, M.; Thompson, T.G.; McPherson, J.D.; Kote-Jarai, Z.; Carpten, J.D.; DiDonato, C.J.; Ikeda, J.E.; Surh, L.; et al. A multicopy dinucleotide marker that maps close to the spinal muscular atrophy gene. Genomics 1994, 21, 394–402. [Google Scholar] [CrossRef]
- Cobben, J.; Van der Steege, G.; Grootscholten, P.; De Visser, M.; Scheffer, H.; Buys, C. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J. Hum. Genet. 1995, 57, 805. [Google Scholar]
- Hahnen, E.; Forkert, R.; Marke, C.; Rudnik-Schöneborn, S.; Schönling, J.; Zerres, K.; Creavin, T.; Wirth, B. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet. 1995, 4, 1927–1933. [Google Scholar] [CrossRef]
- Ackermann, B.; Kröber, S.; Torres-Benito, L.; Borgmann, A.; Peters, M.; Hosseini Barkooie, S.M.; Tejero, R.; Jakubik, M.; Schreml, J.; Milbradt, J.; et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 2013, 22, 1328–1347. [Google Scholar] [CrossRef]
- Heesen, L.; Peitz, M.; Torres-Benito, L.; Hölker, I.; Hupperich, K.; Dobrindt, K.; Jungverdorben, J.; Ritzenhofen, S.; Weykopf, B.; Eckert, D.; et al. Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell. Mol. Life Sci. 2016, 73, 2089–2104. [Google Scholar] [CrossRef] [PubMed]
- Hasanzad, M.; Azad, M.; Kahrizi, K.; Saffar, B.S.; Nafisi, S.; Keyhanidoust, Z.; Azimian, M.; Refah, A.; Urtizberea, J.A.; Tizzano, E.F.; et al. Carrier frequency of SMA by quantitative analysis of the SMN1 deletion in the Iranian population. Eur. J. Neurol. 2010, 17, 160. [Google Scholar] [CrossRef] [PubMed]
- Kaifer, K.A.; Villalón, E.; Osman, E.Y.; Glascock, J.J.; Arnold, L.L.; Cornelison, D.; Lorson, C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2017, 2, e89970. [Google Scholar] [CrossRef] [PubMed]
- McGovern, V.L.; Massoni-Laporte, A.; Wang, X.; Le, T.T.; Le, H.T.; Beattie, C.E.; Rich, M.M.; Burghes, A.H. Plastin 3 expression does not modify spinal muscular atrophy severity in the Δ7 SMA mouse. PLoS ONE 2015, 10, e0132364. [Google Scholar] [CrossRef][Green Version]
- Hao, L.T.; Wolman, M.; Granato, M.; Beattie, C.E. Survival motor neuron affects plastin 3 protein levels leading to motor defects. J. Neurosci. 2012, 32, 5074–5084. [Google Scholar] [CrossRef]
- Janzen, E.; Mendoza-Ferreira, N.; Hosseinibarkooie, S.; Schneider, S.; Hupperich, K.; Tschanz, T.; Grysko, V.; Riessland, M.; Hammerschmidt, M.; Rigo, F.; et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 2018, 141, 2343–2361. [Google Scholar] [CrossRef]
- Torres-Benito, L.; Schneider, S.; Rombo, R.; Ling, K.K.; Grysko, V.; Upadhyay, A.; Kononenko, N.L.; Rigo, F.; Bennett, C.F.; Wirth, B. NCALD Antisense oligonucleotide therapy in addition to nusinersen further ameliorates spinal muscular atrophy in mice. Am. J. Hum. Genet. 2019, 105, 221–230. [Google Scholar] [CrossRef]
- Hassan, H.A.; Zaki, M.S.; Issa, M.Y.; El-Bagoury, N.M.; Essawi, M.L. Genetic pattern of SMN1, SMN2, and NAIP genes in prognosis of SMA patients. Egypt. J. Med. Hum. Genet. 2020, 21, 1–7. [Google Scholar] [CrossRef]
- Essawi, M.; Effat, L.; Shanab, G.; Al-Ettribi, G.; El-Haronui, A.; Karim, A. Molecular analysis of SMN1 and NAIP genes in Egyptian patients with spinal muscular atrophy. Bratislavske lekarske listy 2007, 108, 133. [Google Scholar]
- Hauke, J.; Riessland, M.; Lunke, S.; Eyüpoglu, I.Y.; Blümcke, I.; El-Osta, A.; Wirth, B.; Hahnen, E. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum. Mol. Genet. 2009, 18, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.Y.; Qu, Y.J.; He, S.X.; Li, Y.; Bai, J.L.; Jin, Y.W.; Wang, H.; Song, F. Association between SMN2 methylation and disease severity in Chinese children with spinal muscular atrophy. J. Zhejiang Univ. Sci. B 2016, 17, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyakova, G.Y.; Voisin, S.; Kiselev, A.V.; Almén, M.S.; Xavier, M.J.; Maretina, M.A.; Tishchenko, L.I.; Fredriksson, R.; Baranov, V.S.; Schiöth, H.B. Genome-wide analysis shows association of epigenetic changes in regulators of Rab and Rho GTPases with spinal muscular atrophy severity. Eur. J. Hum. Genet. 2013, 21, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyakova, G.Y.; Nilsson, E.K.; Kiselev, A.V.; Maretina, M.A.; Tishchenko, L.I.; Fredriksson, R.; Baranov, V.S.; Schiöth, H.B. Methylation levels of SLC23A2 and NCOR2 genes correlate with spinal muscular atrophy severity. PLoS ONE 2015, 10, 0121964. [Google Scholar] [CrossRef]
- Aguilera, O.; Fernández, A.F.; Muñoz, A.; Fraga, M.F. Epigenetics and environment: A complex relationship. J. Appl. Physiol. 2010, 109, 243–251. [Google Scholar] [CrossRef]
- Chang, J.G.; Hsieh-Li, H.M.; Jong, Y.J.; Wang, N.M.; Tsai, C.H.; Li, H. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. USA 2001, 98, 9808–9813. [Google Scholar] [CrossRef]
- Brichta, L.; Hofmann, Y.; Hahnen, E.; Siebzehnrubl, F.; Raschke, H.; Blumcke, I.; Eyupoglu, I.; Wirth, B. Valproic acid increases the SMN2 protein level: A well-known drug as a potential therapy for spinal muscular atrophy. Hum. Mol. Genet. 2003, 12, 2481–2489. [Google Scholar] [CrossRef]
- Sumner, C.J.; Huynh, T.N.; Markowitz, J.A.; Perhac, J.S.; Hill, B.; Coovert, D.D.; Schussler, K.; Chen, X.; Jarecki, J.; Burghes, A.H.; et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann.Neurol. Offi. J. Am. Neurol. Assoc. Child Neurol. Soc. 2003, 54, 647–654. [Google Scholar] [CrossRef]
- Hahnen, E.; Eyüpoglu, I.Y.; Brichta, L.; Haastert, K.; Tränkle, C.; Siebzehnrübl, F.A.; Riessland, M.; Hölker, I.; Claus, P.; Romstöck, J.; et al. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J. Neurochem. 2006, 98, 193–202. [Google Scholar] [CrossRef]
- Mohseni, J.; Al-Najjar, B.O.; Wahab, H.A.; Zabidi-Hussin, Z.; Sasongko, T.H. Transcript, methylation and molecular docking analyses of the effects of HDAC inhibitors, SAHA and Dacinostat, on SMN2 expression in fibroblasts of SMA patients. J. Hum. Genet. 2016, 61, 823–830. [Google Scholar] [CrossRef]
- Riessland, M.; Brichta, L.; Hahnen, E.; Wirth, B. The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Hum. Genet. 2006, 120, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.M.; Burnett, B.G.; Taye, A.A.; Gabanella, F.; Knight, M.A.; Hartenstein, P.; Cizman, Z.; Di Prospero, N.A.; Pellizzoni, L.; Fischbeck, K.H.; et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2007, 117, 659–671. [Google Scholar] [CrossRef]
- Garbes, L.; Riessland, M.; Hölker, I.; Heller, R.; Hauke, J.; Tränkle, C.; Coras, R.; Blümcke, I.; Hahnen, E.; Wirth, B. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum. Mol. Genet. 2009, 18, 3645–3658. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Guerra, M.; Di Rosa, V.; Compagnucci, C.; Sette, C. Combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in Spinal Muscular Atrophy cells. J. Neurochem. 2020, 153, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, K.J.; Scott, C.B.; Reyna, S.P.; Prior, T.W.; LaSalle, B.; Sorenson, S.L.; Wood, J.; Acsadi, G.; Crawford, T.O.; Kissel, J.T.; et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS ONE 2009, 4, e5268. [Google Scholar] [CrossRef]
- Swoboda, K.J.; Scott, C.B.; Crawford, T.O.; Simard, L.R.; Reyna, S.P.; Krosschell, K.J.; Acsadi, G.; Elsheik, B.; Schroth, M.K.; D’Anjou, G.; et al. SMA CARNI-VAL trial part I: Double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS ONE 2010, 5, e12140. [Google Scholar] [CrossRef]
- Krosschell, K.J.; Kissel, J.T.; Townsend, E.L.; Simeone, S.D.; Zhang, R.Z.; Reyna, S.P.; Crawford, T.O.; Schroth, M.K.; Acsadi, G.; Kishnani, P.S.; et al. Clinical trial of L-Carnitine and valproic acid in spinal muscular atrophy type I. Muscle Nerve 2018, 57, 193–199. [Google Scholar] [CrossRef]
- Kissel, J.T.; Scott, C.B.; Reyna, S.P.; Crawford, T.O.; Simard, L.R.; Krosschell, K.J.; Acsadi, G.; Elsheik, B.; Schroth, M.K.; D’Anjou, G.; et al. SMA carni-VAL trial part II: A prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS ONE 2011, 6, e21296. [Google Scholar] [CrossRef]
- Kissel, J.T.; Elsheikh, B.; King, W.M.; Freimer, M.; Scott, C.B.; Kolb, S.J.; Reyna, S.P.; Crawford, T.O.; Simard, L.R.; Krosschell, K.J.; et al. SMA valiant trial: A prospective, double-blind, placebo-controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Muscle Nerve 2014, 49, 187–192. [Google Scholar] [CrossRef]
- Darbar, I.A.; Plaggert, P.G.; Resende, M.B.D.; Zanoteli, E.; Reed, U.C. Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid. BMC Neurol. 2011, 11, 36. [Google Scholar] [CrossRef]
- Elshafay, A.; Hieu, T.H.; Doheim, M.F.; Kassem, M.A.M.; ELdoadoa, M.F.; Holloway, S.K.; Abo-Elghar, H.; Hirayama, K.; Huy, N.T. Efficacy and safety of valproic acid for spinal muscular atrophy: A systematic review and meta-analysis. CNS Drugs 2019, 33, 239–250. [Google Scholar] [CrossRef]
- Brahe, C.; Vitali, T.; Tiziano, F.D.; Angelozzi, C.; Pinto, A.M.; Borgo, F.; Moscato, U.; Bertini, E.; Mercuri, E.; Neri, G. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur. J. Hum. Genet. 2005, 13, 256–259. [Google Scholar] [CrossRef]
- Mercuri, E.; Bertini, E.; Messina, S.; Pelliccioni, M.; D’Amico, A.; Colitto, F.; Mirabella, M.; Tiziano, F.D.; Vitali, T.; Angelozzi, C.; et al. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul. Disord. 2004, 14, 130–135. [Google Scholar] [CrossRef]
- Mercuri, E.; Bertini, E.; Messina, S.; Solari, A.; D’amico, A.; Angelozzi, C.; Battini, R.; Berardinelli, A.; Boffi, P.; Bruno, C.; et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 2007, 68, 51–55. [Google Scholar] [CrossRef]
- Liu, Q.; Dreyfuss, G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996, 15, 3555–3565. [Google Scholar] [CrossRef]
- Li, D.K.; Tisdale, S.; Lotti, F.; Pellizzoni, L. SMN control of RNP assembly: From post-transcriptional gene regulation to motor neuron disease. Semin. Cell Dev. Biol. 2014, 32, 22–29. [Google Scholar] [CrossRef]
- Hensel, N.; Claus, P. The actin cytoskeleton in SMA and ALS: How does it contribute to motoneuron degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Donlin-Asp, P.G.; Bassell, G.J.; Rossoll, W. A role for the survival of motor neuron protein in mRNP assembly and transport. Curr. Opin. Neurobiol. 2016, 39, 53–61. [Google Scholar] [CrossRef]
- Groen, E.J.; Gillingwater, T.H. UBA1: At the crossroads of ubiquitin homeostasis and neurodegeneration. Trends Mol. Med. 2015, 21, 622–632. [Google Scholar] [CrossRef]
- Boyd, P.J.; Tu, W.Y.; Shorrock, H.K.; Groen, E.J.; Carter, R.N.; Powis, R.A.; Thomson, S.R.; Thomson, D.; Graham, L.C.; Motyl, A.A.; et al. Bioenergetic status modulates motor neuron vulnerability and pathogenesis in a zebrafish model of spinal muscular atrophy. PLoS Genet. 2017, 13, e1006744. [Google Scholar] [CrossRef]
- Kong, L.; Wang, X.; Choe, D.W.; Polley, M.; Burnett, B.G.; Bosch-Marcé, M.; Griffin, J.W.; Rich, M.M.; Sumner, C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009, 29, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Akten, B.; Kye, M.J.; Hao, L.T.; Wertz, M.H.; Singh, S.; Nie, D.; Huang, J.; Merianda, T.T.; Twiss, J.L.; Beattie, C.E.; et al. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc. Natl. Acad. Sci. USA 2011, 108, 10337–10342. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Zhang, H.; Su, Y.; Silani, V.; Singer, R.H.; Rossoll, W.; Bassell, G.J. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly (A) mRNA in primary motor neuron axons. J. Neurosci. 2011, 31, 3914–3925. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Singh, N.N.; Luo, D.; Singh, R.N. High-affinity RNA targets of the Survival Motor Neuron protein reveal diverse preferences for sequence and structural motifs. Nucleic Acids Res. 2018, 46, 10983–11001. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Besse, A.; Astord, S.; Marais, T.; Roda, M.; Giroux, B.; Lejeune, F.X.; Relaix, F.; Smeriglio, P.; Barkats, M.; Biferi, M.G. AAV9-mediated expression of SMN restricted to neurons does not rescue the spinal muscular atrophy phenotype in mice. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Ramos, D.M.; d’Ydewalle, C.; Gabbeta, V.; Dakka, A.; Klein, S.K.; Norris, D.A.; Matson, J.; Taylor, S.J.; Zaworski, P.G.; Prior, T.W.; et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J. Clin. Investig. 2019, 129, 4817–4831. [Google Scholar] [CrossRef]
- Wadman, R.I.; Stam, M.; Jansen, M.D.; van der Weegen, Y.; Wijngaarde, C.A.; Harschnitz, O.; Sodaar, P.; Braun, K.P.; Dooijes, D.; Lemmink, H.H.; et al. A comparative study of SMN protein and mRNA in blood and fibroblasts in patients with spinal muscular atrophy and healthy controls. PLoS ONE 2016, 11, e0167087. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Petzold, A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.; Veeranna Nixon, R. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.E.; Sternberger, N.H.; Sternberger, L.A. Phosphorylation protects neurofilaments against proteolysis. J. Neuroimmunol. 1987, 14, 149–160. [Google Scholar] [CrossRef]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef]
- Wurster, C.D.; Steinacker, P.; Günther, R.; Koch, J.C.; Lingor, P.; Uzelac, Z.; Witzel, S.; Wollinsky, K.; Winter, B.; Osmanovic, A.; et al. Neurofilament light chain in serum of adolescent and adult SMA patients under treatment with nusinersen. J. Neurol. 2020, 267, 36–44. [Google Scholar] [CrossRef]
- Totzeck, A.; Stolte, B.; Kizina, K.; Bolz, S.; Schlag, M.; Thimm, A.; Kleinschnitz, C.; Hagenacker, T. Neurofilament heavy chain and tau protein are not elevated in cerebrospinal fluid of adult patients with spinal muscular atrophy during loading with nusinersen. Int. J. Mol. Sci. 2019, 20, 5397. [Google Scholar] [CrossRef]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Scarafino, A.; D’Errico, E.; Introna, A.; Fraddosio, A.; Distaso, E.; Tempesta, I.; Morea, A.; Mastronardi, A.; Leante, R.; Ruggieri, M.; et al. Diagnostic and prognostic power of CSF Tau in amyotrophic lateral sclerosis. J. Neurol. 2018, 265, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- Winter, B.; Guenther, R.; Ludolph, A.C.; Hermann, A.; Otto, M.; Wurster, C.D. Neurofilaments and tau in CSF in an infant with SMA type 1 treated with nusinersen. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1068–1069. [Google Scholar] [CrossRef]
- Kim, J.K.; Jha, N.N.; Feng, Z.; Faleiro, M.R.; Chiriboga, C.A.; Wei-Lapierre, L.; Dirksen, R.T.; Ko, C.P.; Monani, U.R. Muscle-specific SMN reduction reveals motor neuron—Independent disease in spinal muscular atrophy models. J. Clin. Investig. 2020, 130, 1271–1287. [Google Scholar] [CrossRef]
- Fayzullina, S.; Martin, L.J. Skeletal muscle DNA damage precedes spinal motor neuron DNA damage in a mouse model of Spinal Muscular Atrophy (SMA). PLoS ONE 2014, 9, e93329. [Google Scholar] [CrossRef]
- Alves, C.R.; Zhang, R.; Johnstone, A.J.; Garner, R.; Nwe, P.H.; Siranosian, J.J.; Swoboda, K.J. Serum creatinine is a biomarker of progressive denervation in spinal muscular atrophy. Neurology 2020, 94, e921–e931. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Crawford, T.O.; Swoboda, K.J.; Kaufmann, P.; Juhasz, P.; Li, X.; Guo, Y.; Li, R.H.; Trachtenberg, F.; Forrest, S.J.; et al. Candidate proteins, metabolites and transcripts in the Biomarkers for Spinal Muscular Atrophy (BforSMA) clinical study. PLoS ONE 2012, 7, e35462. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Soh, B.S.; Rodriguez-Muela, N.; Hendrickson, D.G.; Price, F.; Rinn, J.L.; Rubin, L.L. Genome-wide RNA-Seq of human motor neurons implicates selective ER stress activation in spinal muscular atrophy. Cell Stem Cell 2015, 17, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.; Nizzardo, M.; Vashisht, S.; Molteni, E.; Melzi, V.; Taiana, M.; Salani, S.; Santonicola, P.; Di Schiavi, E.; Bucchia, M.; et al. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain 2019, 142, 276–294. [Google Scholar] [CrossRef]
- Nizzardo, M.; Taiana, M.; Rizzo, F.; Benitez, J.A.; Nijssen, J.; Allodi, I.; Melzi, V.; Bresolin, N.; Comi, G.; Hedlund, E.; et al. Synaptotagmin 13 is neuroprotective across motor neuron diseases. Acta Neuropathol. 2020, 139, 837–853. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef]
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 2018, 24, 335–341. [Google Scholar] [CrossRef]
- Allicock, O.M.; Guo, C.; Uhlemann, A.C.; Whittier, S.; Chauhan, L.V.; Garcia, J.; Price, A.; Morse, S.S.; Mishra, N.; Briese, T.; et al. BacCapSeq: A platform for diagnosis and characterization of bacterial infections. MBio 2018, 9, e02007-18. [Google Scholar] [CrossRef]
- Gibbs, R.M.; Lipnick, S.; Bateman, J.W.; Chen, L.; Cousins, H.C.; Hubbard, E.G.; Jowett, G.; LaPointe, D.S.; McGredy, M.J.; Odonkor, M.N.; et al. Toward precision medicine for neurological and neuropsychiatric disorders. Cell Stem Cell 2018, 23, 21–24. [Google Scholar] [CrossRef]
- Seferian, A.M.; Moraux, A.; Canal, A.; Decostre, V.; Diebate, O.; Le Moing, A.G.; Gidaro, T.; Deconinck, N.; Van Parys, F.; Vereecke, W.; et al. Upper limb evaluation and one-year follow up of non-ambulant patients with spinal muscular atrophy: An observational multicenter trial. PLoS ONE 2015, 10, e0121799. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| SMA Type | Subtype | Age of Onset | Level of Motor Functions | Life Expectancy | SMN2cn (%) |
|---|---|---|---|---|---|
| 0/1a | pre-natal | Need respiratory assistance | <1 month | ||
| 1 | 0–6 mo. | Cannot sit independently | <2 years | 2 (73.4%) | |
| 1b | Absence of head control and ability to roll over | ||||
| 1c | Sometimes gain head control or the ability to roll from supine to prone position | ||||
| 2 | <18 mo. | Cannot stand independently | >2 years | 3 (81.8%) | |
| 2a | Independent sitting lost | ||||
| 2b | Independent sitting conserved | ||||
| 3 | >18 mo. | Able to stand and walk independently | Adulthood | 3–4 (50.6%; 45.5%) | |
| 3a | 18 mo.–3 y. | ||||
| 3b | >3 y. | ||||
| 4 | >20 y. | Weaknesses in lower limbs | Adulthood |
| References | Cohort | T | HFMSE | N | Age |
|---|---|---|---|---|---|
| Natural history studies |  | ||||
| Kaufmann et al. [27] | type 2 | / |  | 41 | 9.1 (7.4) |
| type 3 | / |  | 38 | 13.7 (10.8) | |
| Montes et al. [31] | type 2 | / |  | 67 | 10.9 (8.3) |
| type 3 | / |  | 59 | 13.4 (10.7) | |
| Faravelli et al. [29] | type 3 | / |  | 12 | 29 (15–35) |
| Walter et al. [18] | type 3 | / |  | 19 | 29 (15–35) |
| Kessler et al. [30] | type 3b | 0 |  | 7 | 39 (13) |
| HFMSE () | |||||
| Nusinersen-related studies |  | ||||
| Kessler et al. [30] | type 3b | +10 mo. |  | 7 | 39 (13) |
| Hagenacker et al. [19] | type 2 | +10 mo. |  | 30 | |
| type 3 | +10 mo. |  | 60 | 37 (12) |
| References | Cohort | T | RULM | N | Age |
|---|---|---|---|---|---|
| Natural history studies |  | ||||
| Stolte et al. [36] | type 2 | / |  | 9 | 24 to 48 |
| type 3 | 0 |  | 19 | 18 to 61 | |
| Walter et al. [18] | type 3 | 0 |  | 19 | 18 to 59 |
| RULM () | |||||
| Nusinersen-related study |  | ||||
| Hagenacker et al. [19] | type 2 | +10 mo. |  | 30 | NA |
| type 3 | +10 mo. |  | 58 | NA |
| References | Cohort | T | Distance (m) | N | Age |
|---|---|---|---|---|---|
| Natural history studies |  | ||||
| Mckay et al. [40] | Healthy | / |  | 400 | 20 to 59 |
| Montes et al. [28] | type 3a | 0 |  | 57 | 10.3 (9.8) |
| type 3b | 0 |  | 28 | 25.6 (12.5) | |
| Distance () | |||||
 | |||||
| Montes et al. [28] | type 3a | +1 y |  | 57 | 10.3 (9.8) |
| type 3b | +1 y |  | 28 | 25.6 (12.5) | |
| Nusinersen-related studies |  | ||||
| Hagenacker et al. [19] | type 3 | +6 mo. |  | 47 | NA |
| +10 mo. |  | 37 | NA | ||
| +14 mo. |  | 25 | NA |
| References | -Omics | Samples’ Source | Highlights |
|---|---|---|---|
| Finkel et al. [144] | Metabolomic, transcriptomic, proteomic | Plasma and urine from 108 SMA patients type 1, 2 and 3 (between 2 and 12 years of age) | 97 proteins and 59 metabolites in the plasma together with 44 metabolites in the urine correlated with functional score |
| Rizzo et al. [146] | Transcriptomic | iPSCs-derived motorneurons from SMA patients and healthy controls | NRXN2 protein downregulation was identified as potentially neuroprotective. |
| Nizzardo et al. [147] | Transcriptomic | Spinal and ocular motoneurons isolated from human central nervous system sections from MND patients | Synaptogamin13 was identified as a putative neuroprotective protein in MND. |
| Kessler et al. [30] | Proteomic | CSF samples from 10 Nusinersen-treated adults SMA type 2 and 3 | No correlation between protein profiling and functional score evolution, over 10 months treatment |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smeriglio, P.; Langard, P.; Querin, G.; Biferi, M.G. The Identification of Novel Biomarkers Is Required to Improve Adult SMA Patient Stratification, Diagnosis and Treatment. J. Pers. Med. 2020, 10, 75. https://doi.org/10.3390/jpm10030075
Smeriglio P, Langard P, Querin G, Biferi MG. The Identification of Novel Biomarkers Is Required to Improve Adult SMA Patient Stratification, Diagnosis and Treatment. Journal of Personalized Medicine. 2020; 10(3):75. https://doi.org/10.3390/jpm10030075
Chicago/Turabian StyleSmeriglio, Piera, Paul Langard, Giorgia Querin, and Maria Grazia Biferi. 2020. "The Identification of Novel Biomarkers Is Required to Improve Adult SMA Patient Stratification, Diagnosis and Treatment" Journal of Personalized Medicine 10, no. 3: 75. https://doi.org/10.3390/jpm10030075
APA StyleSmeriglio, P., Langard, P., Querin, G., & Biferi, M. G. (2020). The Identification of Novel Biomarkers Is Required to Improve Adult SMA Patient Stratification, Diagnosis and Treatment. Journal of Personalized Medicine, 10(3), 75. https://doi.org/10.3390/jpm10030075