A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS

 , ,
, ,  and
and 
Abstract
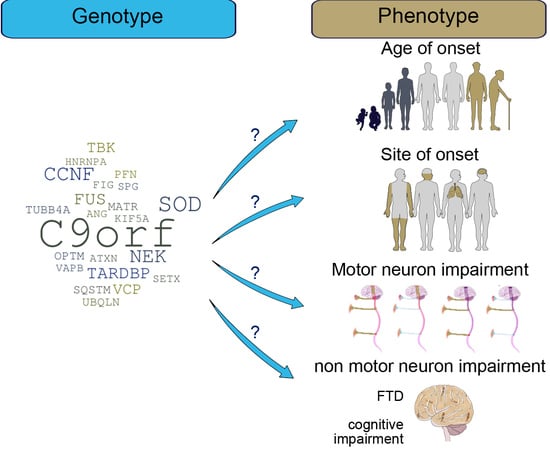
1. Introduction
2. Pathological Definition of ALS: Clinical Features and Phenotype Variability
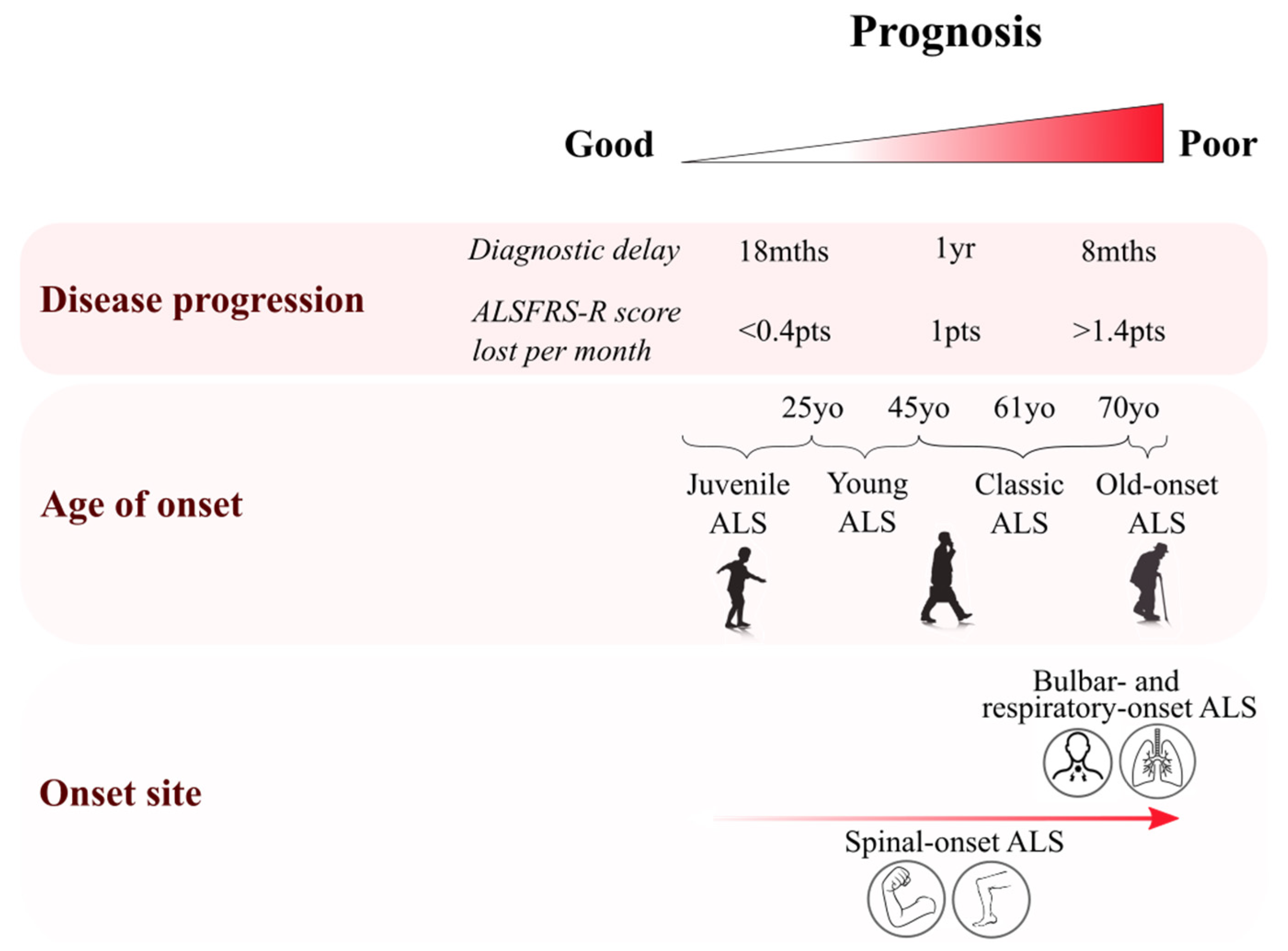
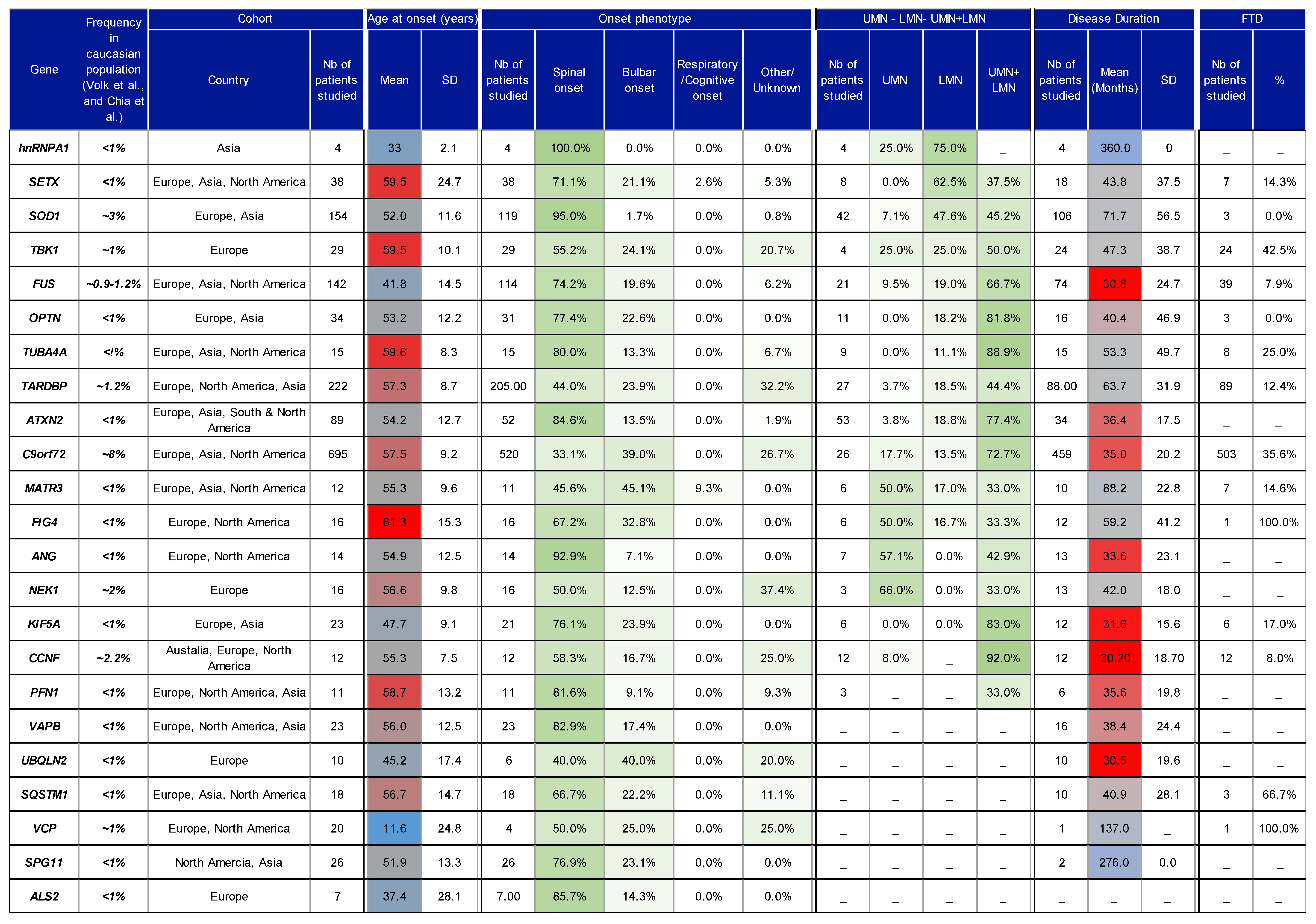
2.1. Age of Onset Variation
2.2. Site of Onset Variability
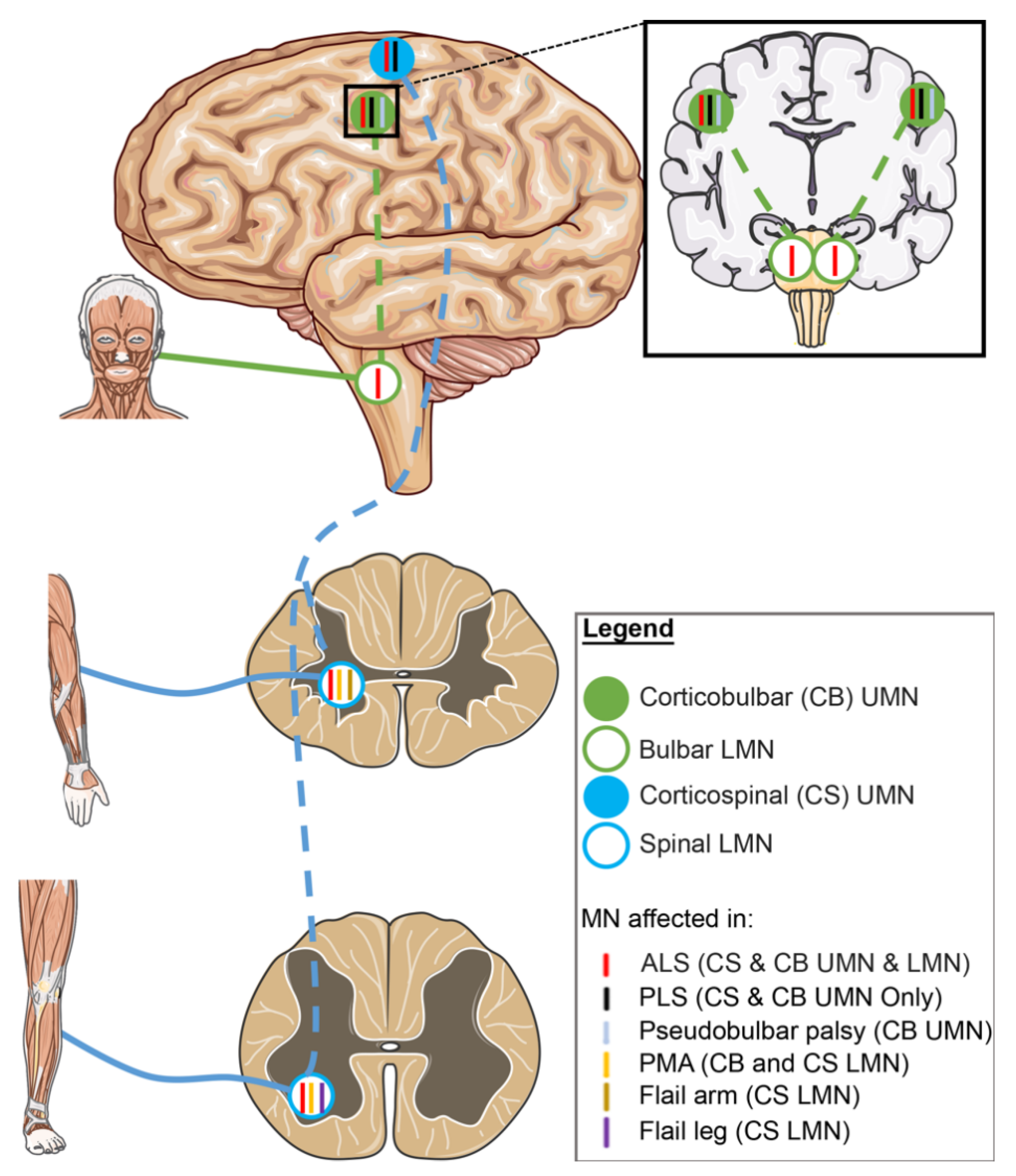
2.3. Motor Neuron Involvement in ALS Variants
2.3.1. UMN-Dominant ALS Variants
2.3.2. LMN-Dominant ALS Variants
2.4. Non-Motor Involvement in ALS and Overlap with FTD
2.4.1. Dementia in ALS Patients—ALS-FTD Variants
2.4.2. Cognitive Changes in Non-Demented ALS Patients—ALSci and ALSbi Variants
3. Genetics of ALS
4. Correlation of Genotype/Phenotype: Methods, Results and Discussion
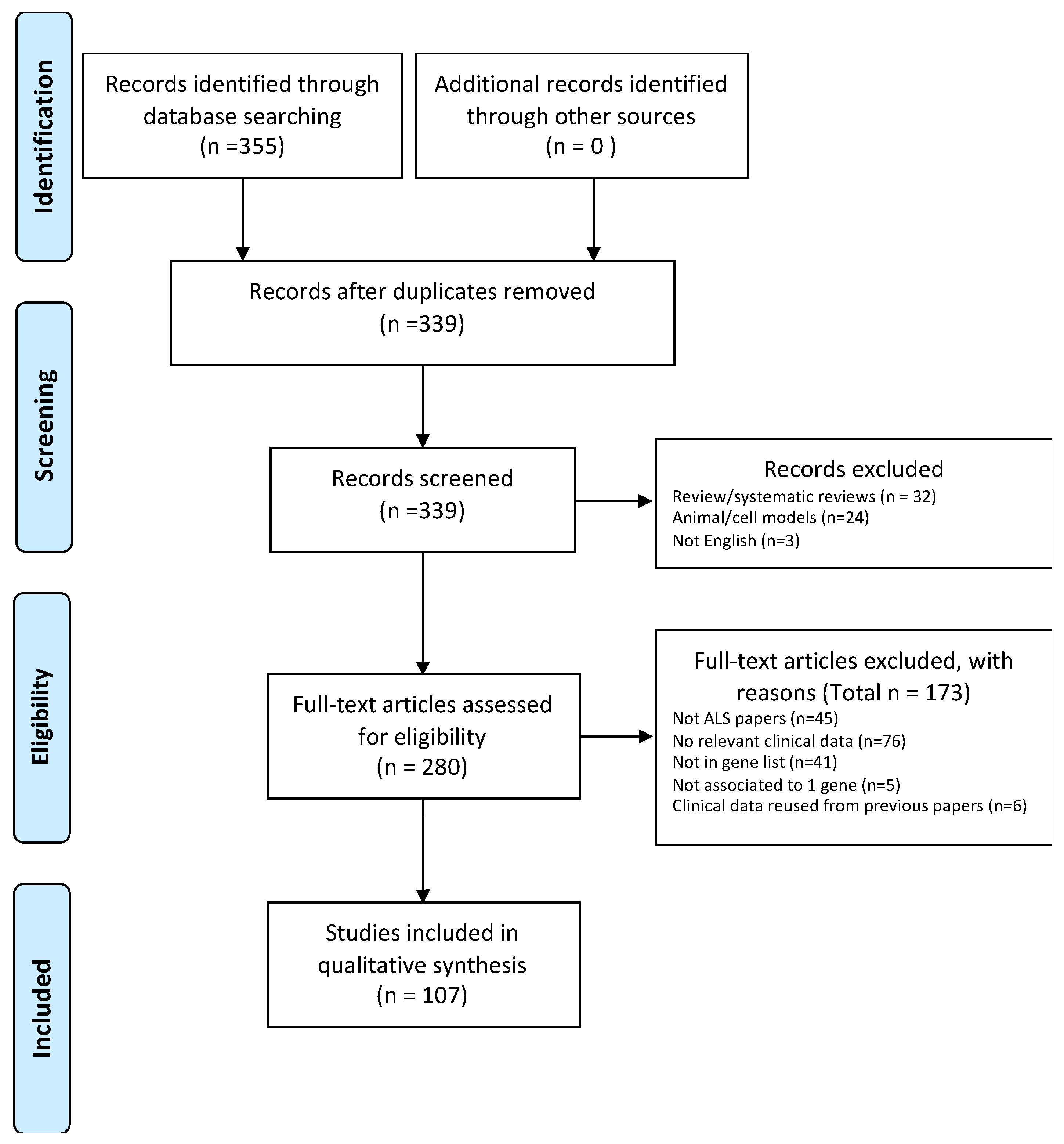
4.1. Protocol
4.2. Eligibility Criteria
4.3. Data Extractions and Synthesis
4.4. Characteristics of Studies
4.5. Overall Findings and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Logroscino, G.; Piccininni, M.; Marin, B.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Alahdab, F.; Asgedom, S.W.; Awasthi, A.; Chaiah, Y.; et al. Global, regional, and national burden of motor neuron diseases 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 1083–1097. [Google Scholar] [CrossRef]
- Gordon, P.H. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 138, pp. 225–238. [Google Scholar]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef]
- Mehta, P.; Kaye, W.; Raymond, J.; Wu, R.; Larson, T.; Punjani, R.; Heller, D.; Cohen, J.; Peters, T.; Muravov, O.; et al. Prevalence of Amyotrophic Lateral Sclerosis—United States. Morb. Mortal. Wkly. Rep. 2014, 67, 216. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef]
- Salameh, J.; Brown, R.; Berry, J. Amyotrophic Lateral Sclerosis: Review. Semin. Neurol. 2015, 35, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Antel, J.; Bradley, W.; Cardy, P.; Carpenter, S.; Chou, S.; Conradi, S.; Daube, J.; Denys, E.H.; Festoff, B.; et al. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J. Neurol. Sci. 1994, 124, 96–107. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; Vasta, R.; Brunetti, M.; Barberis, M.; Corrado, L.; D’Alfonso, S.; Bersano, E.; et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 2019, 93, e984–e994. [Google Scholar] [CrossRef]
- Forbes, R.B.; Colville, S.; Swingler, R.J.; Scottish ALS/MND Register. The epidemiology of amyotrophic lateral sclerosis (ALS/MND) in people aged 80 or over. Age Ageing 2004, 33, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; van den Berg, L.H. Amyotrophic lateral sclerosis: Moving towards a new classification system. Lancet Neurol. 2016, 15, 1182–1194. [Google Scholar] [CrossRef]
- Vijayakumar, U.G.; Milla, V.; Cynthia Stafford, M.Y.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.-R. A Systematic Review of Suggested Molecular Strata, Biomarkers and Their Tissue Sources in ALS. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Cutler, D.J.; Yarab, N.; Kelly, C.M.; Glass, J.D. The Heritability of Amyotrophic Lateral Sclerosis in a Clinically Ascertained United States Research Registry. PLoS ONE 2011, 6, e27985. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G. Le Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. medizinische Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Yoshida, M.; Takahashi, Y.; Koike, A.; Fukuda, Y.; Goto, J.; Tsuji, S. A mutation database for amyotrophic lateral sclerosis. Hum. Mutat. 2010, 31, 1003–1010. [Google Scholar] [CrossRef]
- McCann, E.P.; Williams, K.L.; Fifita, J.A.; Tarr, I.S.; O’Connor, J.; Rowe, D.B.; Nicholson, G.A.; Blair, I.P. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin. Genet. 2017, 92, 259–266. [Google Scholar] [CrossRef]
- Li, H.-F.; Wu, Z.-Y. Genotype-phenotype correlations of amyotrophic lateral sclerosis. Transl. Neurodegener. 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, M.; Conte, A.; Zollino, M. Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin. Genet. 2013, 83, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Salachas, F.; Cazeneuve, C.; Gordon, P.; Bricka, B.; Camuzat, A.; Guillot-Noël, L.; Russaouen, O.; Bruneteau, G.; Pradat, P.-F.; et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Turner, M.R.; Barnwell, J.; Al-Chalabi, A.; Eisen, A. Young-onset amyotrophic lateral sclerosis: Historical and other observations. Brain 2012, 135, 2883–2891. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. On Behalf of the Eurals Consortium Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef]
- Sabatelli, M.; Madia, F.; Conte, A.; Luigetti, M.; Zollino, M.; Mancuso, I.; Lo Monaco, M.; Lippi, G.; Tonali, P. Natural history of young-adult amyotrophic lateral sclerosis. Neurology 2008, 71, 876–881. [Google Scholar] [CrossRef]
- Tard, C.; Defebvre, L.; Moreau, C.; Devos, D.; Danel-Brunaud, V. Clinical features of amyotrophic lateral sclerosis and their prognostic value. Rev. Neurol. 2017, 173, 263–272. [Google Scholar] [CrossRef]
- Garden, G.A.; La Spada, A.R. Intercellular (mis)communication in neurodegenerative disease. Neuron 2012, 73, 886–901. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Bäumer, D.; Talbot, K.; Turner, M.R. Advances in motor neurone disease. J. R. Soc. Med. 2014, 107, 14–21. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Lillo, P.; Hodges, J.R. Frontotemporal dementia and motor neurone disease: Overlapping clinic-pathological disorders. J. Clin. Neurosci. 2009, 16, 1131–1135. [Google Scholar] [CrossRef]
- D’Amico, E.; Pasmantier, M.; Lee, Y.-W.; Weimer, L.; Mitsumoto, H. Clinical evolution of pure upper motor neuron disease/dysfunction (PUMMD). Muscle Nerve 2013, 47, 28–32. [Google Scholar] [CrossRef]
- Statland, J.M.; Barohn, R.J.; McVey, A.L.; Katz, J.S.; Dimachkie, M.M. Patterns of Weakness, Classification of Motor Neuron Disease, and Clinical Diagnosis of Sporadic Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve 2010, 41, 161–165. [Google Scholar] [CrossRef]
- Riku, Y.; Atsuta, N.; Yoshida, M.; Tatsumi, S.; Iwasaki, Y.; Mimuro, M.; Watanabe, H.; Ito, M.; Senda, J.; Nakamura, R.; et al. Differential motor neuron involvement in progressive muscular atrophy: A comparative study with amyotrophic lateral sclerosis. BMJ Open 2014, 4, e005213. [Google Scholar] [CrossRef]
- Liewluck, T.; Saperstein, D.S. Progressive Muscular Atrophy. Neurol. Clin. 2015, 33, 761–773. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Mathers, S.; Talman, P.; Galtrey, C.; Parkinson, M.H.; Ganesalingam, J.; Willey, E.; Ampong, M.A.; Ellis, C.M.; Shaw, C.E.; et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology 2009, 72, 1087–1094. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Chen, S.-D.; Feng, S.-Y.; Liu, C.-Y.; Cui, M.; Chen, S.; Feng, S.-M.; Dong, Q.; Huang, H.; Yu, J.-T. Familial flail leg ALS caused by PFN1 mutation. J. Neurol. Neurosurg. Psychiatry 2020, 91, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Park, S.B.; Vucic, S.; Yiannikas, C.; Spies, J.; Howells, J.; Huynh, W.; Matamala, J.M.; Krishnan, A.V.; Pollard, J.D.; et al. Differentiating lower motor neuron syndromes. J. Neurol. Neurosurg. Psychiatry 2017, 88, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.H.; Abrahams, S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: Nature of impairment and implications for assessment. Lancet Neurol. 2013, 12, 368–380. [Google Scholar] [CrossRef]
- Shen, D.; Cui, L.; Fang, J.; Cui, B.; Li, D.; Tai, H. Voxel-Wise Meta-Analysis of Gray Matter Changes in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2016, 8. [Google Scholar] [CrossRef]
- Geser, F.; Brandmeir, N.J.; Kwong, L.K.; Martinez-Lage, M.; Elman, L.; McCluskey, L.; Xie, S.X.; Lee, V.M.-Y.; Trojanowski, J.Q. Evidence of Multisystem Disorder in Whole-Brain Map of Pathological TDP-43 in Amyotrophic Lateral Sclerosis. Arch. Neurol. 2008, 65. [Google Scholar] [CrossRef]
- Crockford, C.; Newton, J.; Lonergan, K.; Chiwera, T.; Booth, T.; Chandran, S.; Colville, S.; Heverin, M.; Mays, I.; Pal, S.; et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 2018, 91, e1370–e1380. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef]
- Bak, T.H.; Chandran, S. What wires together dies together: Verbs, actions and neurodegeneration in motor neuron disease. Cortex 2012, 48, 936–944. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; Mclaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; HortobáGyi, T.; et al. Amyotrophic lateral sclerosis—Frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Yang, Y.; Hentati, A.; Deng, H.-X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.-Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Sheerin, U.-M.; Schneider, S.A.; Carr, L.; Deuschl, G.; Hopfner, F.; Stamelou, M.; Wood, N.W.; Bhatia, K.P. ALS2 mutations: Juvenile amyotrophic lateral sclerosis and generalized dystonia. Neurology 2014, 82, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-Onset Ascending Hereditary Spastic Paralysis Is Associated with Mutations in the Alsin Gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Otomo, A. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.; Spannl, S.; Ferguson, C.; Hyman, A.A.; Parton, R.G.; Zerial, M. Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. Elife 2018, 7. [Google Scholar] [CrossRef]
- Devon, R.S.; Orban, P.C.; Gerrow, K.; Barbieri, M.A.; Schwab, C.; Cao, L.P.; Helm, J.R.; Bissada, N.; Cruz-Aguado, R.; Davidson, T.-L.; et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 9595–9600. [Google Scholar] [CrossRef]
- Cai, H. Loss of ALS2 Function Is Insufficient to Trigger Motor Neuron Degeneration in Knock-Out Mice But Predisposes Neurons to Oxidative Stress. J. Neurosci. 2005, 25, 7567–7574. [Google Scholar] [CrossRef]
- Suraweera, A.; Lim, Y.; Woods, R.; Birrell, G.W.; Nasim, T.; Becherel, O.J.; Lavin, M.F. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum. Mol. Genet. 2009, 18, 3384–3396. [Google Scholar] [CrossRef]
- Steinmetz, E.J.; Warren, C.L.; Kuehner, J.N.; Panbehi, B.; Ansari, A.Z.; Brow, D.A. Genome-Wide Distribution of Yeast RNA Polymerase II and Its Control by Sen1 Helicase. Mol. Cell 2006, 24, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA Helicase Gene Mutations in a Form of Juvenile Amyotrophic Lateral Sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Avemaria, F.; Lunetta, C.; Tarlarini, C.; Mosca, L.; Maestri, E.; Marocchi, A.; Melazzini, M.; Penco, S.; Corbo, M. Mutation in the senataxin gene found in a patient affected by familial ALS with juvenile onset and slow progression. Amyotroph. Lateral Scler. 2011, 12, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Arning, L.; Epplen, J.T.; Zerres, K. SETX gene mutation in a family diagnosed autosomal dominant proximal spinal muscular atrophy. Neuromuscul. Disord. 2012, 22, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yu, W.; Kishikawa, H.; Folkerth, R.D.; Iafrate, A.J.; Shen, Y.; Xin, W.; Sims, K.; Hu, G. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann. Neurol. 2007, 62, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Crabtree, B.; Acharya, K.R. Human angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/pathfinding and survival of motor neurons. Hum. Mol. Genet. 2007, 17, 130–149. [Google Scholar] [CrossRef]
- Tsuji, T.; Sun, Y.; Kishimoto, K.; Olson, K.A.; Liu, S.; Hirukawa, S.; Hu, G. Angiogenin Is Translocated to the Nucleus of HeLa Cells and Is Involved in Ribosomal RNA Transcription and Cell Proliferation. Cancer Res. 2005, 65, 1352–1360. [Google Scholar] [CrossRef]
- Thiyagarajan, N.; Ferguson, R.; Subramanian, V.; Acharya, K.R. Structural and molecular insights into the mechanism of action of human angiogenin-ALS variants in neurons. Nat. Commun. 2012, 3, 1121. [Google Scholar] [CrossRef]
- Kanekura, K.; Nishimoto, I.; Aiso, S.; Matsuoka, M. Characterization of Amyotrophic Lateral Sclerosis-linked P56S Mutation of Vesicle-associated Membrane Protein-associated Protein B (VAPB/ALS8). J. Biol. Chem. 2006, 281, 30223–30233. [Google Scholar] [CrossRef]
- Peretti, D.; Dahan, N.; Shimoni, E.; Hirschberg, K.; Lev, S. Coordinated Lipid Transfer between the Endoplasmic Reticulum and the Golgi Complex Requires the VAP Proteins and Is Essential for Golgi-mediated Transport. Mol. Biol. Cell 2008, 19, 3871–3884. [Google Scholar] [CrossRef]
- Tsuda, H.; Han, S.M.; Yang, Y.; Tong, C.; Lin, Y.Q.; Mohan, K.; Haueter, C.; Zoghbi, A.; Harati, Y.; Kwan, J.; et al. The Amyotrophic Lateral Sclerosis 8 Protein VAPB Is Cleaved, Secreted, and Acts as a Ligand for Eph Receptors. Cell 2008, 133, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Morotz, G.M.; De Vos, K.J.; Vagnoni, A.; Ackerley, S.; Shaw, C.E.; Miller, C.C.J. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 2012, 21, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Chalhoub, A.; Schooley, A.; Zhang, W.; Ngsee, J.K. A mutation in VAPB that causes amyotrophic lateral sclerosis also causes a nuclear envelope defect. J. Cell Sci. 2012, 125, 2831–2836. [Google Scholar] [CrossRef]
- Qiu, L.; Qiao, T.; Beers, M.; Tan, W.; Wang, H.; Yang, B.; Xu, Z. Widespread aggregation of mutant VAPB associated with ALS does not cause motor neuron degeneration or modulate mutant SOD1 aggregation and toxicity in mice. Mol. Neurodegener. 2013, 8, 1. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Fujioka, S.; Traynor, S.; Soto-Ortolaza, A.I.; Petrucelli, L.; Dickson, D.W.; Rademakers, R.; Boylan, K.B.; Graff-Radford, N.R.; Uitti, R.J.; et al. TARDBP mutations in Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 312–315. [Google Scholar] [CrossRef]
- Schwab, C.; Arai, T.; Hasegawa, M.; Yu, S.; McGeer, P.L. Colocalization of Transactivation-Responsive DNA-Binding Protein 43 and Huntingtin in Inclusions of Huntington Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Sweeney, F.; Bodi, I.; Troakes, C.; Maekawa, S.; Al-Sarraj, S. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer’s disease. Neuropathology 2010, 30, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Nakashima-Yasuda, H.; Uryu, K.; Robinson, J.; Xie, S.X.; Hurtig, H.; Duda, J.E.; Arnold, S.E.; Siderowf, A.; Grossman, M.; Leverenz, J.B.; et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007, 114, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious Variants of FIG4, a Phosphoinositide Phosphatase, in Patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; et al. Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in Sarcoma (FUS) Protein Lacking Nuclear Localization Signal (NLS) and Major RNA Binding Motifs Triggers Proteinopathy and Severe Motor Phenotype in Transgenic Mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
- Gentile, F.; Scarlino, S.; Falzone, Y.M.; Lunetta, C.; Tremolizzo, L.; Quattrini, A.; Riva, N. The Peripheral Nervous System in Amyotrophic Lateral Sclerosis: Opportunities for Translational Research. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.E.; Li, J.; Zhang, X.; et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef]
- Tsai, C.-P.; Soong, B.-W.; Lin, K.-P.; Tu, P.-H.; Lin, J.-L.; Lee, Y.-C. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol. Aging 2011, 32, 553.e13–553.e21. [Google Scholar] [CrossRef] [PubMed]
- Verdiani, S.; Origone, P.; Geroldi, A.; Bandettini Di Poggio, M.; Mantero, V.; Bellone, E.; Mancardi, G.; Caponnetto, C.; Mandich, P. The FIG4 gene does not play a major role in causing ALS in Italian patients. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 228–229. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Shen, L.; Du, J.; Zhao, G.; Wang, X.; Yang, Y.; Xiao, Z.; Yuan, Y.; Jiang, H.; Li, N.; et al. Novel mutations of the SPG11 gene in hereditary spastic paraplegia with thin corpus callosum. J. Neurol. Sci. 2008, 275, 92–99. [Google Scholar] [CrossRef]
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Fittschen, M.; Lastres-Becker, I.; Halbach, M.V.; Damrath, E.; Gispert, S.; Azizov, M.; Walter, M.; Müller, S.; Auburger, G. Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate. Neurogenetics 2015, 16, 181–192. [Google Scholar] [CrossRef]
- Satterfield, T.F.; Pallanck, L.J. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum. Mol. Genet. 2006, 15, 2523–2532. [Google Scholar] [CrossRef]
- Van Damme, P.; Veldink, J.H.; van Blitterswijk, M.; Corveleyn, A.; van Vught, P.W.J.; Thijs, V.; Dubois, B.; Matthijs, G.; van den Berg, L.H.; Robberecht, W. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 2011, 76, 2066–2072. [Google Scholar] [CrossRef]
- Liu, X.; Lu, M.; Tang, L.; Zhang, N.; Chui, D.; Fan, D. ATXN2 CAG repeat expansions increase the risk for Chinese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 2236.e5–2236.e8. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Oania, R.S.; Kolawa, N.J.; Deshaies, R.J. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife 2013, 2. [Google Scholar] [CrossRef]
- Rumpf, S.; Bagley, J.A.; Thompson-Peer, K.L.; Zhu, S.; Gorczyca, D.; Beckstead, R.B.; Jan, L.Y.; Jan, Y.N. Drosophila Valosin-Containing Protein is required for dendrite pruning through a regulatory role in mRNA metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 7331–7336. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Yin, H.Z.; Nalbandian, A.; Hsu, C.-I.; Li, S.; Llewellyn, K.J.; Mozaffar, T.; Kimonis, V.E.; Weiss, J.H. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 2012, 3, e374. [Google Scholar] [CrossRef]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3–2527.e10. [Google Scholar] [CrossRef]
- Goutman, S.A.; Chen, K.S.; Paez-Colasante, X.; Feldman, E.L. Emerging understanding of the genotype–phenotype relationship in amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2018, 148, 603–623. [Google Scholar]
- Mizuno, Y.; Amari, M.; Takatama, M.; Aizawa, H.; Mihara, B.; Okamoto, K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006, 249, 13–18. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.R.; Boeve, B.F.F.; Boxer, A.L.L.; Baker, M.; Rutherford, N.J.J.; Nicholson, A.M.M.; Finch, N.A.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Umoh, M.E.; Fournier, C.; Li, Y.; Polak, M.; Shaw, L.; Landers, J.E.; Hu, W.; Gearing, M.; Glass, J.D. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016, 87, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; Fogh, I.; et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Frolov, A.; Highley, J.R.; Charlesworth, G.; Kirby, J.; Milano, A.; Hartley, J.; Ince, P.G.; McDermott, C.J.; Lashley, T.; et al. C9ORF72 expansions, parkinsonism, and Parkinson disease: A clinicopathologic study. Neurology 2013, 81, 808–811. [Google Scholar] [CrossRef]
- Devenney, E.M.; Ahmed, R.M.; Halliday, G.; Piguet, O.; Kiernan, M.C.; Hodges, J.R. Psychiatric disorders in C9orf72 kindreds. Neurology 2018, 91, e1498–e1507. [Google Scholar] [CrossRef]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Yang, C.; Danielson, E.W.; Qiao, T.; Metterville, J.; Brown, R.H.; Landers, J.E.; Xu, Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc. Natl. Acad. Sci. USA 2016, 113, E6209–E6218. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Honda, H.; Hamasaki, H.; Wakamiya, T.; Koyama, S.; Suzuki, S.O.; Fujii, N.; Iwaki, T. Loss of hnRNPA1 in ALS spinal cord motor neurons with TDP-43-positive inclusions. Neuropathology 2015, 35, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide Rare Variant Analysis Identifies TUBA4A Mutations Associated with Familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Nguyen, H.P.; Van Mossevelde, S.; Moisse, M.; Sieben, A.; Santens, P.; De Bleecker, J.; Vandenbulcke, M.; Engelborghs, S.; Baets, J.; et al. Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol. Aging 2017, 51, 177.e9–177.e16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, J.; Tang, L.; Chen, L.; Ma, Y.; Fan, D. Screening for TUBA4A mutations in a large Chinese cohort of patients with ALS: Re-evaluating the pathogenesis of TUBA4A in ALS. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1350–1352. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef]
- Salton, M.; Elkon, R.; Borodina, T.; Davydov, A.; Yaspo, M.-L.; Halperin, E.; Shiloh, Y. Matrin 3 Binds and Stabilizes mRNA. PLoS ONE 2011, 6, e23882. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.B.; Attig, J.; Bellora, N.; König, J.; Hallegger, M.; Kayikci, M.; Eyras, E.; Ule, J.; Smith, C.W. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J. 2015, 34, 653–668. [Google Scholar] [CrossRef]
- Boehringer, A.; Garcia-Mansfield, K.; Singh, G.; Bakkar, N.; Pirrotte, P.; Bowser, R. ALS Associated Mutations in Matrin 3 Alter Protein-Protein Interactions and Impede mRNA Nuclear Export. Sci. Rep. 2017, 7, 14529. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Brenner, D.; Müller, K.; Wieland, T.; Weydt, P.; Böhm, S.; Lulé, D.; Hübers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef]
- Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef]
- Yu, Y.; Nakagawa, T.; Morohoshi, A.; Nakagawa, M.; Ishida, N.; Suzuki, N.; Aoki, M.; Nakayama, K. Pathogenic mutations in the ALS gene CCNF cause cytoplasmic mislocalization of Cyclin F and elevated VCP ATPase activity. Hum. Mol. Genet. 2019, 28, 3486–3497. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef]
- Campbell, P.D.; Shen, K.; Sapio, M.R.; Glenn, T.D.; Talbot, W.S.; Marlow, F.L. Unique Function of Kinesin Kif5A in Localization of Mitochondria in Axons. J. Neurosci. 2014, 34, 14717–14732. [Google Scholar] [CrossRef]
- Hancock, W.O.; Howard, J. Processivity of the Motor Protein Kinesin Requires Two Heads. J. Cell Biol. 1998, 140, 1395–1405. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Laura, M.; Hersheson, J.; Horga, A.; Jaunmuktane, Z.; Brandner, S.; Pittman, A.; Hughes, D.; Polke, J.M.; Sweeney, M.G.; et al. Extended phenotypic spectrum of KIF5A mutations: From spastic paraplegia to axonal neuropathy. Neurology 2014, 83, 612–619. [Google Scholar] [CrossRef]
- Gu, X.; Li, C.; Chen, Y.; Wei, Q.; Cao, B.; Ou, R.; Yuan, X.; Hou, Y.; Zhang, L.; Liu, H.; et al. Mutation screening of the KIF5A gene in Chinese patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 245–246. [Google Scholar] [CrossRef]
- Filosto, M.; Piccinelli, S.; Palmieri, I.; Necchini, N.; Valente, M.; Zanella, I.; Biasiotto, G.; Lorenzo, D.; Cereda, C.; Padovani, A. A Novel Mutation in the Stalk Domain of KIF5A Causes a Slowly Progressive Atypical Motor Syndrome. J. Clin. Med. 2018, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Yilmaz, R.; Müller, K.; Grehl, T.; Petri, S.; Meyer, T.; Grosskreutz, J.; Weydt, P.; Ruf, W.; Neuwirth, C.; et al. Hot-spot KIF5A mutations cause familial ALS. Brain 2018, 141, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Faber, I.; Martinez, A.R.M.; de Rezende, T.J.R.; Martins, C.R.; Martins, M.P.; Lourenço, C.M.; Marques, W.; Montecchiani, C.; Orlacchio, A.; Pedroso, J.L.; et al. SPG11 mutations cause widespread white matter and basal ganglia abnormalities, but restricted cortical damage. NeuroImage Clin. 2018, 19, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Lin, H.-X.; Liu, G.-L.; Tao, Q.-Q.; Ni, W.; Xiao, B.-G.; Wu, Z.-Y. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Cui, L.-Y.; Sun, Q.; Li, X.-G.; Liu, M.-S.; Xu, Y.; Zhou, Y.; Yang, X.-Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Ansorge, O.; Strong, M.; Bilbao, J.; Zinman, L.; Ang, L.-C.; Baker, M.; Stewart, H.; Eisen, A.; Rademakers, R.; et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: Two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011, 122, 87–98. [Google Scholar] [CrossRef]
- Waibel, S.; Neumann, M.; Rosenbohm, A.; Birve, A.; Volk, A.E.; Weishaupt, J.H.; Meyer, T.; Müller, U.; Andersen, P.M.; Ludolph, A.C. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: A clinico-genetic study in Germany. Eur. J. Neurol. 2013, 20, 540–546. [Google Scholar] [CrossRef]
- Orban, P.; Devon, R.S.; Hayden, M.R.; Leavitt, B.R. Chapter 15 Juvenile amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2007; pp. 301–312. [Google Scholar]
- Zhao, Z.; Chen, W.; Wu, Z.; Wang, N.; Zhao, G.; Chen, W.; Murong, S. A novel mutation in the senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 118–122. [Google Scholar] [CrossRef]
- Murphy, N.A.; Arthur, K.C.; Tienari, P.J.; Houlden, H.; Chiò, A.; Traynor, B.J. Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep. 2017, 7, 2116. [Google Scholar] [CrossRef]
- Trojsi, F.; Siciliano, M.; Femiano, C.; Santangelo, G.; Lunetta, C.; Calvo, A.; Moglia, C.; Marinou, K.; Ticozzi, N.; Ferro, C.; et al. Comparative Analysis of C9orf72 and Sporadic Disease in a Large Multicenter ALS Population: The Effect of Male Sex on Survival of C9orf72 Positive Patients. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Rooney, J.; Fogh, I.; Westeneng, H.-J.; Vajda, A.; McLaughlin, R.; Heverin, M.; Jones, A.; van Eijk, R.; Calvo, A.; Mazzini, L.; et al. C9orf72 expansion differentially affects males with spinal onset amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 281. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.X.; Edwards, B.; Lee, S.; Finelli, M.J.; Davies, B.; Davies, K.E.; Oliver, P.L. Neuron-specific antioxidant OXR1 extends survival of a mouse model of amyotrophic lateral sclerosis. Brain 2015, 138, 1167–1181. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Grüneberg, M.; Seelhöfer, A.; et al. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain 2019, 142, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Tasca, G.; Lattante, S.; Marangi, G.; Conte, A.; Bernardo, D.; Bisogni, G.; Mandich, P.; Zollino, M.; Ragozzino, E.; Udd, B.; et al. SOD1 p.D12Y variant is associated with ALS/distal myopathy spectrum. Eur. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stewart, H.; Rutherford, N.J.; Briemberg, H.; Krieger, C.; Cashman, N.; Fabros, M.; Baker, M.; Fok, A.; DeJesus-Hernandez, M.; Eisen, A.; et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 2012, 123, 409–417. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Canosa, A.; Calvo, A.; Moglia, C.; Barberis, M.; Brunetti, M.; Cammarosano, S.; Manera, U.; Ilardi, A.; Restagno, G.; Chiò, A. A novel p.E121G heterozygous missense mutation of SOD1 in an apparently sporadic ALS case with a 14-year course. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 127–128. [Google Scholar] [CrossRef]
- Corcia, P.; Vourc’h, P.; Blasco, H.; Couratier, P.; Dangoumau, A.; Bellance, R.; Desnuelle, C.; Viader, F.; Pautot, V.; Millecamps, S.; et al. Phenotypic and genotypic studies of ALS cases in ALS-SMA families. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 432–437. [Google Scholar] [CrossRef]
- Battistini, S.; Ricci, C.; Giannini, F.; Calzavara, S.; Greco, G.; Del Corona, A.; Mancuso, M.; Battistini, N.; Siciliano, G.; Carrera, P. G41S SOD1 mutation: A common ancestor for six ALS Italian families with an aggressive phenotype. Amyotroph. Lateral Scler. 2010, 11, 210–215. [Google Scholar] [CrossRef]
- Nizzardo, M.; Simone, C.; Rizzo, F.; Ulzi, G.; Ramirez, A.; Rizzuti, M.; Bordoni, A.; Bucchia, M.; Gatti, S.; Bresolin, N.; et al. Morpholino-mediated SOD1 reduction ameliorates an amyotrophic lateral sclerosis disease phenotype. Sci. Rep. 2016, 6, 21301. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Isaacs, A.M.; Mizielinska, S.; Mead, S.; Lashley, T.; Wray, S.; Sidle, K.; Fratta, P.; Orrell, R.W.; Hardy, J.; et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. 2015, 14, 291–301. [Google Scholar] [CrossRef]
- Le Ber, I. SQSTM1 Mutations in French Patients With Frontotemporal Dementia or Frontotemporal Dementia With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Rohrer, J.D.; Real, R.; Lubbe, S.J.; Waite, A.J.; Blake, D.J.; Walters, R.J.; Lashley, T.; Revesz, T.; Holton, J.L.; et al. A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Mol. Case Stud. 2019, 5, a003913. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Wilke, C.; Simón-Sánchez, J.; Jansen, I.E.; Reifschneider, A.; Capell, A.; Haass, C.; Castillo-Lizardo, M.; Biskup, S.; Maetzler, W.; et al. The wide genetic landscape of clinical frontotemporal dementia: Systematic combined sequencing of 121 consecutive subjects. Genet. Med. 2018, 20, 240–249. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Liu, Q.; Shu, S.; Wang, R.R.; Liu, F.; Cui, B.; Guo, X.N.; Lu, C.X.; Li, X.G.; Liu, M.S.; Peng, B.; et al. Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS. Neurology 2016, 87, 1763–1769. [Google Scholar] [CrossRef]
- Tripolszki, K.; Török, D.; Goudenège, D.; Farkas, K.; Sulák, A.; Török, N.; Engelhardt, J.I.; Klivényi, P.; Procaccio, V.; Nagy, N.; et al. High-throughput sequencing revealed a novel SETX mutation in a Hungarian patient with amyotrophic lateral sclerosis. Brain Behav. 2017, 7, e00669. [Google Scholar] [CrossRef]
- Kenna, K.P.; McLaughlin, R.L.; Byrne, S.; Elamin, M.; Heverin, M.; Kenny, E.M.; Cormican, P.; Morris, D.W.; Donaghy, C.G.; Bradley, D.G.; et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J. Med. Genet. 2013, 50, 776–783. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Lin, H.-X.; Wei, Q.; Zhang, Q.-J.; Chen, C.-X.; Tao, Q.-Q.; Liu, G.-L.; Ni, W.; Gitler, A.D.; Li, H.-F.; et al. Genetic Spectrum and Variability in Chinese Patients with Amyotrophic Lateral Sclerosis. Aging Dis. 2019, 10, 1199. [Google Scholar] [CrossRef]
- Tsai, Y.; Lin, K.; Jih, K.; Tsai, P.; Liao, Y.; Lee, Y. Hand-onset weakness is a common feature of ALS patients with a NEK1 loss-of-function variant. Ann. Clin. Transl. Neurol. 2020, acn3.51064. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Giannini, F.; Intini, E.; Battistini, S. Genotype–phenotype correlation and evidence for a common ancestor in two Italian ALS patients with the D124G SOD1 mutation. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Dalla Bella, E.; Lombardi, R.; Porretta-Serapiglia, C.; Ciano, C.; Gellera, C.; Pensato, V.; Cazzato, D.; Lauria, G. Amyotrophic lateral sclerosis causes small fiber pathology. Eur. J. Neurol. 2016, 23, 416–420. [Google Scholar] [CrossRef]
- Khani, M.; Alavi, A.; Nafissi, S.; Elahi, E. Observation of c.260A > G mutation in superoxide dismutase 1 that causes p.Asn86Ser in Iranian amyotrophic lateral sclerosis patient and absence of genotype/phenotype correlation. Iran. J. Neurol. 2015, 14, 152–157. [Google Scholar] [PubMed]
- Chiò, A.; Mora, G.; Sabatelli, M.; Caponnetto, C.; Lunetta, C.; Traynor, B.J.; Johnson, J.O.; Nalls, M.A.; Calvo, A.; Moglia, C.; et al. HFE p.H63D polymorphism does not influence ALS phenotype and survival. Neurobiol. Aging 2015, 36, 2906.e7–2906.e11. [Google Scholar]
- Kim, M.-J.; Bae, J.-H.; Kim, J.-M.; Kim, H.R.; Yoon, B.-N.; Sung, J.-J.; Ahn, S.-W. Rapid Progression of Sporadic ALS in a Patient Carrying SOD1 p.Gly13Arg Mutation. Exp. Neurobiol. 2016, 25, 347–350. [Google Scholar] [CrossRef]
- Chen, W.; Xie, Y.; Zheng, M.; Lin, J.; Huang, P.; Pei, Z.; Yao, X. Clinical and genetic features of patients with amyotrophic lateral sclerosis in southern China. Eur. J. Neurol. 2020, 27, 1017–1022. [Google Scholar] [CrossRef]
- Lattante, S.; Conte, A.; Zollino, M.; Luigetti, M.; Del Grande, A.; Marangi, G.; Romano, A.; Marcaccio, A.; Meleo, E.; Bisogni, G.; et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 2012, 79, 66–72. [Google Scholar] [CrossRef]
- Felbecker, A.; Camu, W.; Valdmanis, P.N.; Sperfeld, A.D.; Waibel, S.; Steinbach, P.; Rouleau, G.A.; Ludolph, A.C.; Andersen, P.M. Four familial ALS pedigrees discordant for two SOD1 mutations: Are all SOD1 mutations pathogenic? J. Neurol. Neurosurg. Psychiatry 2010, 81, 572–577. [Google Scholar] [CrossRef]
- Nogales-Gadea, G.; Garcia-Arumi, E.; Andreu, A.L.; Cervera, C.; Gamez, J. A novel exon 5 mutation (N139H) in the SOD1 gene in a Spanish family associated with incomplete penetrance. J. Neurol. Sci. 2004, 219, 1–6. [Google Scholar] [CrossRef]
- Ferrera, L.; Caponnetto, C.; Marini, V.; Rizzi, D.; Bordo, D.; Penco, S.; Amoroso, A.; Origone, P.; Garrè, C. An Italian dominant FALS Leu144Phe SOD1 mutation: Genotype-phenotype correlation. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Luisa Conforti, F.; Sprovieri, T.; Mazzei, R.; Patitucci, A.; Ungaro, C.; Zoccolella, S.; Magariello, A.; Bella, V.L.; Tessitore, A.; Tedeschi, G.; et al. Further evidence that D90A-SOD1 mutation is recessively inherited in ALS patients in Italy. Amyotroph. Lateral Scler. 2009, 10, 58–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murakami, T.; Warita, H.; Hayashi, T.; Sato, K.; Manabe, Y.; Mizuno, S.; Yamane, K.; Abe, K. A novel SOD1 gene mutation in familial ALS with low penetrance in females. J. Neurol. Sci. 2001, 189, 45–47. [Google Scholar] [CrossRef]
- Segovia-Silvestre, T.; Andreu, A.L.; Vives-Bauza, C.; Garcia-Arumi, E.; Cervera, C.; Gamez, J. A novel exon 3 mutation (D76V) in the SOD1 gene associated with slowly progressive ALS. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2002, 3, 69–74. [Google Scholar] [CrossRef]
- Ticozzi, N.; Tiloca, C.; Mencacci, N.E.; Morelli, C.; Doretti, A.; Rusconi, D.; Colombrita, C.; Sangalli, D.; Verde, F.; Finelli, P.; et al. Oligoclonal bands in the cerebrospinal fluid of amyotrophic lateral sclerosis patients with disease-associated mutations. J. Neurol. 2013, 260, 85–92. [Google Scholar] [CrossRef]
- Mandrioli, J.; Michalke, B.; Solovyev, N.; Grill, P.; Violi, F.; Lunetta, C.; Conte, A.; Sansone, V.A.; Sabatelli, M.; Vinceti, M. Elevated Levels of Selenium Species in Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients with Disease-Associated Gene Mutations. Neurodegener. Dis. 2017, 17, 171–180. [Google Scholar] [CrossRef]
- van der Zee, J.; Gijselinck, I.; Van Mossevelde, S.; Perrone, F.; Dillen, L.; Heeman, B.; Bäumer, V.; Engelborghs, S.; De Bleecker, J.; Baets, J.; et al. TBK1 Mutation Spectrum in an Extended European Patient Cohort with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Hum. Mutat. 2017, 38, 297–309. [Google Scholar] [CrossRef]
- Weinreich, M.; Shepheard, S.R.; Verber, N.; Wyles, M.; Heath, P.R.; Highley, J.R.; Kirby, J.; Shaw, P.J. Neuropathological characterization of a novel TANK binding kinase (TBK1) gene loss of function mutation associated with amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2019, nan.12578. [Google Scholar] [CrossRef]
- Caroppo, P.; Camuzat, A.; De Septenville, A.; Couratier, P.; Lacomblez, L.; Auriacombe, S.; Flabeau, O.; Jornéa, L.; Blanc, F.; Sellal, F.; et al. Semantic and nonfluent aphasic variants, secondarily associated with amyotrophic lateral sclerosis, are predominant frontotemporal lobar degeneration phenotypes in TBK1 carriers. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit. 2015, 1, 481–486. [Google Scholar] [CrossRef]
- Dols-Icardo, O.; García-Redondo, A.; Rojas-García, R.; Borrego-Hernández, D.; Illán-Gala, I.; Muñoz-Blanco, J.L.; Rábano, A.; Cervera-Carles, L.; Juárez-Rufián, A.; Spataro, N.; et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J. Neurol. Neurosurg. Psychiatry 2018, 89, 162–168. [Google Scholar] [CrossRef]
- Calvo, A.; Moglia, C.; Canosa, A.; Brunetti, M.; Barberis, M.; Traynor, B.J.; Carrara, G.; Valentini, C.; Restagno, G.; Chiò, A. A de novo nonsense mutation of the FUS gene in an apparently familial amyotrophic lateral sclerosis case. Neurobiol. Aging 2014, 35, 1513.e7–1513.e11. [Google Scholar] [CrossRef] [PubMed]
- Damme, P.V.; Goris, A.; Race, V.; Hersmus, N.; Dubois, B.; Bosch, L.V.D.; Matthijs, G.; Robberecht, W. The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur. J. Neurol. 2010, 17, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Liu, M.-S.; Li, X.-G.; Cui, L.-Y. Mutations in FUS are the most frequent genetic cause in juvenile sporadic ALS patients of Chinese origin. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Peikert, K.; Günther, R.; Kooi, A.J.; Aronica, E.; Hübers, A.; Danel, V.; Corcia, P.; Pan-Montojo, F.; Cirak, S.; et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2384–2394. [Google Scholar] [CrossRef]
- Tümer, Z.; Bertelsen, B.; Gredal, O.; Magyari, M.; Nielsen, K.C.; LuCamp; Grønskov, K.; Brøndum-Nielsen, K. A novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol. Aging 2012, 33, 208.e1–208.e5. [Google Scholar]
- Iida, A.; Hosono, N.; Sano, M.; Kamei, T.; Oshima, S.; Tokuda, T.; Nakajima, M.; Kubo, M.; Nakamura, Y.; Ikegawa, S. Novel deletion mutations of OPTN in amyotrophic lateral sclerosis in Japanese. Neurobiol. Aging 2012, 33, 1843.e19–1843.e24. [Google Scholar] [CrossRef]
- Feng, S.; Che, C.; Feng, S.; Liu, C.; Li, L.; Li, Y.; Huang, H.; Zou, Z. Novel mutation in optineurin causing aggressive ALS+/−frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 2377–2383. [Google Scholar] [CrossRef]
- Li, C.; Ji, Y.; Tang, L.; Zhang, N.; He, J.; Ye, S.; Liu, X.; Fan, D. Optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in China. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 485–489. [Google Scholar] [CrossRef]
- Goldstein, O.; Nayshool, O.; Nefussy, B.; Traynor, B.J.; Renton, A.E.; Gana-Weisz, M.; Drory, V.E.; Orr-Urtreger, A. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology 2016, 86, 446–453. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Waibel, S.; Birve, A.; Volk, A.E.; Mayer, B.; Meyer, T.; Ludolph, A.C.; Andersen, P.M. A novel optineurin truncating mutation and three glaucoma-associated missense variants in patients with familial amyotrophic lateral sclerosis in Germany. Neurobiol. Aging 2013, 34, 1516.e9–1516.e15. [Google Scholar] [CrossRef]
- Del Bo, R.; Tiloca, C.; Pensato, V.; Corrado, L.; Ratti, A.; Ticozzi, N.; Corti, S.; Castellotti, B.; Mazzini, L.; Soraru, G.; et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, J.; Tang, L.; Chen, L.; Xu, L.; Ma, Y.; Zhang, N.; Fan, D. TUBA4A may not be a significant genetic factor in Chinese ALS patients. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Pensato, V.; Tiloca, C.; Corrado, L.; Bertolin, C.; Sardone, V.; Del Bo, R.; Calini, D.; Mandrioli, J.; Lauria, G.; Mazzini, L.; et al. TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: Identification of novel mutations. J. Neurol. 2015, 262, 1376–1378. [Google Scholar] [CrossRef]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Occhineri, P.; Cau, T.B.; et al. Genetic architecture of ALS in Sardinia. Neurobiol. Aging 2014, 35, 2882.e7–2882.e12. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology 2012, 78, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Orrù, S.; Manolakos, E.; Orrù, N.; Kokotas, H.; Mascia, V.; Carcassi, C.; Petersen, M.B. High frequency of the TARDBP p.Ala382Thr mutation in Sardinian patients with amyotrophic lateral sclerosis. Clin. Genet. 2012, 81, 172–178. [Google Scholar] [CrossRef]
- Ticozzi, N.; LeClerc, A.L.; van Blitterswijk, M.; Keagle, P.; McKenna-Yasek, D.M.; Sapp, P.C.; Silani, V.; Wills, A.-M.; Brown, R.H.; Landers, J.E. Mutational analysis of TARDBP in neurodegenerative diseases. Neurobiol. Aging 2011, 32, 2096–2099. [Google Scholar] [CrossRef]
- Xu, G.; Hu, W.; Zhan, L.-L.; Wang, C.; Xu, L.-Q.; Lin, M.-T.; Chen, W.-J.; Wang, N.; Zhang, Q.-J. High frequency of the TARDBP p.M337 V mutation among south-eastern Chinese patients with familial amyotrophic lateral sclerosis. BMC Neurol. 2018, 18, 35. [Google Scholar] [CrossRef]
- Caroppo, P.; Camuzat, A.; Guillot-Noel, L.; Thomas-Antérion, C.; Couratier, P.; Wong, T.H.; Teichmann, M.; Golfier, V.; Auriacombe, S.; Belliard, S.; et al. Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol. Genet. 2016, 2, e80. [Google Scholar] [CrossRef]
- Corrado, L.; Mazzini, L.; Oggioni, G.D.; Luciano, B.; Godi, M.; Brusco, A.; D’Alfonso, S. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum. Genet. 2011, 130, 575–580. [Google Scholar] [CrossRef]
- Tavares de Andrade, H.M.; Cintra, V.P.; de Albuquerque, M.; Piccinin, C.C.; Bonadia, L.C.; Duarte Couteiro, R.E.; Sabino de Oliveira, D.; Claudino, R.; Magno Gonçalves, M.V.; Dourado, M.E.T.; et al. Intermediate-length CAG repeat in ATXN2 is associated with increased risk for amyotrophic lateral sclerosis in Brazilian patients. Neurobiol. Aging 2018, 69, 292.e15–292.e18. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Rutherford, N.J.; Baker, M.; Soto-Ortolaza, A.I.; Carrasquillo, M.M.; DeJesus-Hernandez, M.; Adamson, J.; Li, M.; Volkening, K.; Finger, E.; et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 2011, 20, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Boillée, S.; Le Ber, I.; Seilhean, D.; Teyssou, E.; Giraudeau, M.; Moigneu, C.; Vandenberghe, N.; Danel-Brunaud, V.; Corcia, P.; et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J. Med. Genet. 2012, 49, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Zinman, L.; Grinberg, Y.; Moreno, D.; Sato, C.; Bilbao, J.M.; Ghani, M.; Hernández, I.; Ruiz, A.; Boada, M.; et al. Investigation of C9orf72 in 4 Neurodegenerative Disorders. Arch. Neurol. 2012, 69, 1583. [Google Scholar] [CrossRef]
- Dols-Icardo, O.; Garcia-Redondo, A.; Rojas-Garcia, R.; Sanchez-Valle, R.; Noguera, A.; Gomez-Tortosa, E.; Pastor, P.; Hernandez, I.; Esteban-Perez, J.; Suarez-Calvet, M.; et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2014, 23, 749–754. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Niemantsverdriet, E.; Murray, M.E.; Heckman, M.G.; Diehl, N.N.; Brown, P.H.; Baker, M.C.; Finch, N.A.; Bauer, P.O.; et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): A cross-sectional cohort study. Lancet Neurol. 2013, 12, 978–988. [Google Scholar] [CrossRef]
- Gijselinck, I.; Van Langenhove, T.; van der Zee, J.; Sleegers, K.; Philtjens, S.; Kleinberger, G.; Janssens, J.; Bettens, K.; Van Cauwenberghe, C.; Pereson, S.; et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65. [Google Scholar] [CrossRef]
- Goldstein, O.; Gana-Weisz, M.; Nefussy, B.; Vainer, B.; Nayshool, O.; Bar-Shira, A.; Traynor, B.J.; Drory, V.E.; Orr-Urtreger, A. High frequency of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis patients from two founder populations sharing the same risk haplotype. Neurobiol. Aging 2018, 64, 160.e1–160.e7. [Google Scholar] [CrossRef]
- Fournier, C.; Barbier, M.; Camuzat, A.; Anquetil, V.; Lattante, S.; Clot, F.; Cazeneuve, C.; Rinaldi, D.; Couratier, P.; Deramecourt, V.; et al. Relations between C9orf72 expansion size in blood, age at onset, age at collection and transmission across generations in patients and presymptomatic carriers. Neurobiol. Aging 2019, 74, 234.e1–234.e8. [Google Scholar] [CrossRef]
- Zhang, M.; Tartaglia, M.C.; Moreno, D.; Sato, C.; McKeever, P.; Weichert, A.; Keith, J.; Robertson, J.; Zinman, L.; Rogaeva, E. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathol. 2017, 134, 271–279. [Google Scholar] [CrossRef]
- Gendron, T.F.; van Blitterswijk, M.; Bieniek, K.F.; Daughrity, L.M.; Jiang, J.; Rush, B.K.; Pedraza, O.; Lucas, J.A.; Murray, M.E.; Desaro, P.; et al. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. 2015, 130, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Kaivorinne, A.-L.; Bode, M.K.; Paavola, L.; Tuominen, H.; Kallio, M.; Renton, A.E.; Traynor, B.J.; Moilanen, V.; Remes, A.M. Clinical Characteristics of C9ORF72-Linked Frontotemporal Lobar Degeneration. Dement. Geriatr. Cogn. Dis. Extra 2013, 3, 251–262. [Google Scholar] [CrossRef] [PubMed]
- van der Burgh, H.K.; Westeneng, H.-J.; Walhout, R.; van Veenhuijzen, K.; Tan, H.H.G.; Meier, J.M.; Bakker, L.A.; Hendrikse, J.; van Es, M.A.; Veldink, J.H.; et al. Multimodal longitudinal study of structural brain involvement in amyotrophic lateral sclerosis. Neurology 2020. [Google Scholar] [CrossRef] [PubMed]
- Floris, G.; Borghero, G.; Di Stefano, F.; Melis, R.; Puddu, R.; Fadda, L.; Murru, M.R.; Corongiu, D.; Cuccu, S.; Tranquilli, S.; et al. Phenotypic variability related to C9orf72 mutation in a large Sardinian kindred. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 245–248. [Google Scholar] [CrossRef]
- Narain, P.; Padhi, A.K.; Dave, U.; Mishra, D.; Bhatia, R.; Vivekanandan, P.; Gomes, J. Identification and characterization of novel and rare susceptible variants in Indian amyotrophic lateral sclerosis patients. Neurogenetics 2019, 20, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Origone, P.; Verdiani, S.; Bandettini Di Poggio, M.; Zuccarino, R.; Vignolo, M.; Caponnetto, C.; Mandich, P. A novel Arg147Trp MATR3 missense mutation in a slowly progressive ALS Italian patient. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 530–531. [Google Scholar] [CrossRef]
- Leblond, C.S.; Gan-Or, Z.; Spiegelman, D.; Laurent, S.B.; Szuto, A.; Hodgkinson, A.; Dionne-Laporte, A.; Provencher, P.; de Carvalho, M.; Orrù, S.; et al. Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2016, 37, 209.e17–209.e21. [Google Scholar] [CrossRef]
- Lin, K.P.; Tsai, P.C.; Liao, Y.C.; Chen, W.T.; Tsai, C.P.; Soong, B.W.; Lee, Y.C. Mutational analysis of MATR3 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 2005.e1–2005.e4. [Google Scholar] [CrossRef]
- Marangi, G.; Lattante, S.; Doronzio, P.N.; Conte, A.; Tasca, G.; Monforte, M.; Patanella, A.K.; Bisogni, G.; Meleo, E.; La Spada, S.; et al. Matrin 3 variants are frequent in Italian ALS patients. Neurobiol. Aging 2017, 49, 218.e1–218.e7. [Google Scholar] [CrossRef]
- Osmanovic, A.; Rangnau, I.; Kosfeld, A.; Abdulla, S.; Janssen, C.; Auber, B.; Raab, P.; Preller, M.; Petri, S.; Weber, R.G. FIG4 variants in central European patients with amyotrophic lateral sclerosis: A whole-exome and targeted sequencing study. Eur. J. Hum. Genet. 2017, 25, 324–331. [Google Scholar] [CrossRef]
- Conforti, F.L.; Sprovieri, T.; Mazzei, R.; Ungaro, C.; La Bella, V.; Tessitore, A.; Patitucci, A.; Magariello, A.; Gabriele, A.L.; Tedeschi, G.; et al. A novel Angiogenin gene mutation in a sporadic patient with amyotrophic lateral sclerosis from southern Italy. Neuromuscul. Disord. 2008, 18, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Paubel, A. Mutations of the ANG Gene in French Patients With Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2008, 65, 1333. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Santiago, R.; Hoenig, S.; Lichtner, P.; Sperfeld, A.-D.; Sharma, M.; Berg, D.; Weichenrieder, O.; Illig, T.; Eger, K.; Meyer, T.; et al. Identification of novel Angiogenin (ANG) gene missense variants in German patients with amyotrophic lateral sclerosis. J. Neurol. 2009, 256, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Van Mossevelde, S.; Dillen, L.; De Bleecker, J.L.; Moisse, M.; Van Damme, P.; Van Broeckhoven, C.; van der Zee, J.; Engelborghs, S.; Crols, R.; et al. NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol. Aging 2018, 61, 255.e1–255.e7. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, X.; Tang, L.; Zhao, C.; He, J.; Fan, D. Whole-exome sequencing identified novel KIF5A mutations in Chinese patients with amyotrophic lateral sclerosis and Charcot-Marie-Tooth type 2. J. Neurol. Neurosurg. Psychiatry 2020, 91, 326–328. [Google Scholar] [CrossRef]
- Tiloca, C.; Ticozzi, N.; Pensato, V.; Corrado, L.; Del Bo, R.; Bertolin, C.; Fenoglio, C.; Gagliardi, S.; Calini, D.; Lauria, G.; et al. Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol. Aging 2013, 34, 1517.e9–1517.e10. [Google Scholar] [CrossRef]
- Ingre, C.; Landers, J.E.; Rizik, N.; Volk, A.E.; Akimoto, C.; Birve, A.; Hübers, A.; Keagle, P.J.; Piotrowska, K.; Press, R.; et al. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol. Aging 2013, 34, 1708.e1–1708.e6. [Google Scholar] [CrossRef]
- Smith, B.N.; Vance, C.; Scotter, E.L.; Troakes, C.; Wong, C.H.; Topp, S.; Maekawa, S.; King, A.; Mitchell, J.C.; Lund, K.; et al. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 2015, 36, 1602.e17–1602.e27. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Z.-Z.; Huang, R.; Chen, K.; Song, W.; Zhao, B.; Chen, X.; Yang, Y.; Yuan, L.; Shang, H.-F. PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 1922.e1–1922.e5. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; van Es, M.A.; Koppers, M.; van Rheenen, W.; Medic, J.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol. Aging 2012, 33, 2950.e1–2950.e4. [Google Scholar] [CrossRef]
- Di, L.; Chen, H.; Da, Y.; Wang, S.; Shen, X.-M. Atypical familial amyotrophic lateral sclerosis with initial symptoms of pain or tremor in a Chinese family harboring VAPB-P56S mutation. J. Neurol. 2016, 263, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Gellera, C.; Tiloca, C.; Del Bo, R.; Corrado, L.; Pensato, V.; Agostini, J.; Cereda, C.; Ratti, A.; Castellotti, B.; Corti, S.; et al. Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2013, 84, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Dillen, L.; Van Langenhove, T.; Engelborghs, S.; Vandenbulcke, M.; Sarafov, S.; Tournev, I.; Merlin, C.; Cras, P.; Vandenberghe, R.; De Deyn, P.P.; et al. Explorative genetic study of UBQLN2 and PFN1 in an extended Flanders-Belgian cohort of frontotemporal lobar degeneration patients. Neurobiol. Aging 2013, 34, 1711.e1–1711.e5. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tang, L.; Zhang, N.; Pan, L.; Hadano, S.; Fan, D. Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Al-Obeidi, E.; Al-Tahan, S.; Surampalli, A.; Goyal, N.; Wang, A.K.; Hermann, A.; Omizo, M.; Smith, C.; Mozaffar, T.; Kimonis, V. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin. Genet. 2018, 93, 119–125. [Google Scholar] [CrossRef]
- Daoud, H.; Zhou, S.; Noreau, A.; Sabbagh, M.; Belzil, V.; Dionne-Laporte, A.; Tranchant, C.; Dion, P.; Rouleau, G.A. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol. Aging 2012, 33, 839.e5–839.e9. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Moll, T.; Ramesh, T.; Castelli, L.; Beer, A.; Robins, H.; Fox, I.; Niedermoser, I.; Van Damme, P.; Moisse, M.; et al. Mutations in the Glycosyltransferase Domain of GLT8D1 Are Associated with Familial Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 26, 2298–2306.e5. [Google Scholar] [CrossRef] [PubMed]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia—Amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Farhan, S.M.K.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; McKenna-Yasek, D.; Sapp, P.E.; Chin, W.; Geller, B.; Hayden, D.L.; Schoenfeld, D.A.; Hosler, B.A.; Horvitz, H.R.; Brown, R.H. Epidemiology of mutations in superoxide dismutase in amyotrophic lateal sclerosis. Ann. Neurol. 1997, 41, 210–221. [Google Scholar] [CrossRef]
- Ogasawara, M.; Matsubara, Y.; Narisawa, K.; Aoki, M.; Nakamura, S.; Itoyama, Y.; Abe, K. Mild ALS in Japan associated with novel SOD mutation. Nat. Genet. 1993, 5, 323–324. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Juvenile | Young | Mid–Late Adulthood | Elderly | |
|---|---|---|---|---|
| Age of onset | ≤25 years old | 25 to 45 years old | 45 to 70 years old | >70 years old |
| M:F ratio | _ | 3–3.6:1 | 1.3–1.56:1 | 1:1.25 |
| Genetics | Mostly familial cases (FUS, SETX, ALS2 mutations) | Mostly familial | ~90% sALS ~10% fALS | _ |
| Site of onset: | ||||
| Limb onset | _ | _ | ~70% | ~40% |
| Bulbar onset | _ | ~16% | ~25% | ~50% (M:F = 1:1.6) |
| Respiratory/cognitive onset | _ | _ | ~5% | _ |
| Survival (from symptoms onset) | Generally longer survival > 10 years | Variable: 50%: <30 months 5–10%: 5–10 years | ~20 months | |
| MN involvement: | ||||
| UMN+LMN | _ | ~40% | ~80% | ~72% |
| UMN-predominant | Predominant | ~60% | ~17% | _ |
| LMN-predominant | _ | _ | _ | ~19% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58. https://doi.org/10.3390/jpm10030058
Connolly O, Le Gall L, McCluskey G, Donaghy CG, Duddy WJ, Duguez S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. Journal of Personalized Medicine. 2020; 10(3):58. https://doi.org/10.3390/jpm10030058
Chicago/Turabian StyleConnolly, Owen, Laura Le Gall, Gavin McCluskey, Colette G Donaghy, William J Duddy, and Stephanie Duguez. 2020. "A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS" Journal of Personalized Medicine 10, no. 3: 58. https://doi.org/10.3390/jpm10030058
APA StyleConnolly, O., Le Gall, L., McCluskey, G., Donaghy, C. G., Duddy, W. J., & Duguez, S. (2020). A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. Journal of Personalized Medicine, 10(3), 58. https://doi.org/10.3390/jpm10030058